ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
ฮามาร์โตม่า
ตรวจสอบล่าสุด: 29.06.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
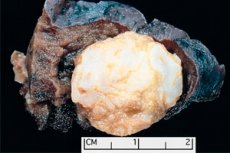
การก่อตัวของเนื้องอกที่เกิดขึ้นในบริเวณกายวิภาคใดๆ อันเป็นผลจากการเติบโตผิดปกติของเนื้อเยื่อที่ไม่ร้ายแรง ในทางการแพทย์เรียกว่า hamartoma (จากภาษากรีก hamartia ซึ่งแปลว่า ข้อผิดพลาด ข้อบกพร่อง) [ 1 ]
ระบาดวิทยา
ตามสถิติแล้ว hamartoma คิดเป็น 1.2% ของเนื้องอกชนิดไม่ร้ายแรง อุบัติการณ์ของ hamartoma ในปอดอยู่ที่ประมาณ 0.25% ของประชากรทั่วไปและคิดเป็น 8% ของเนื้องอกในปอดทั้งหมด hamartoma ในปอดส่วนใหญ่มักได้รับการวินิจฉัยโดยบังเอิญในผู้ป่วยที่มีอายุ 40 ถึง 70 ปี แต่พบได้น้อยมากในทางการแพทย์เด็ก
โดยทั่วไปแล้ว hamartomas ส่วนใหญ่ได้รับการวินิจฉัยในผู้ชาย ถึงแม้ว่า hamartomas ที่ไตจะพบได้บ่อยในผู้หญิงมากกว่า และมักจะตรวจพบในวัยกลางคน
ประมาณ 5% ของเนื้องอกเต้านมชนิดไม่ร้ายแรงคือเนื้องอกชนิด hamartoma และส่วนใหญ่มักเกิดขึ้นกับผู้หญิงที่มีอายุมากกว่า 35 ปี
ร้อยละ 80-90 ของรอยโรค hamartomatous ของสมองและร้อยละ 50 ของรอยโรค hamartomatous ของหัวใจมีความเกี่ยวข้องกับโรค Tuberous Sclerosis
สาเหตุ ของฮามาร์โทมา
Gamartomas เป็นความผิดปกติแต่กำเนิดและมีลักษณะไม่ร้ายแรง ซึ่งเกิดจากเนื้อเยื่อมีเซนไคมอลที่มาจากแผ่นเซลล์สืบพันธุ์ และสาเหตุของการเกิดขึ้นนั้นเกี่ยวข้องกับการแบ่งเซลล์ที่ไม่สามารถควบคุมได้ของเนื้อเยื่อปกติทางเซลล์ (เนื้อเยื่อเกี่ยวพัน กล้ามเนื้อเรียบ ไขมัน หรือกระดูกอ่อน) ซึ่งเป็นลักษณะเฉพาะของตำแหน่งทางกายวิภาคที่กำหนด และการเจริญเติบโตมากเกินไปเฉพาะจุดในระหว่างการสร้างตัวอ่อนของอวัยวะหรือโครงสร้างทางกายวิภาคเกือบทุกประเภท
การเกิด hamartomas หลายอันในผู้ป่วยรายเดียวกัน มักเรียกกันว่า hamartomatosis หรือ pleiotropic hamartoma
เนื้องอกเหล่านี้อาจเกิดขึ้นเป็นครั้งคราวหรือในภาวะที่มีโรคทางพันธุกรรมแบบถ่ายทอดลักษณะทางพันธุกรรมบางชนิด รวมถึงกลุ่มอาการที่กำหนดโดยพันธุกรรมด้วย
ในหลายกรณี hamartomas เกิดขึ้นเมื่อโรคทางพันธุกรรมที่หายากซึ่งมีลักษณะหลายระบบ - tuberous sclerosis - ปรากฏขึ้นในเวลาไม่นานหลังคลอด หรือในโรคทางกรรมพันธุ์ของ Recklinghausen - neurofibromatosisชนิดที่ 1 [ 2 ]
ปัจจัยเสี่ยง
ปัจจัยเสี่ยงหลักต่อการเกิด hamartoma ได้แก่ การมีกลุ่มอาการทางพันธุกรรมที่เรียกว่า hamartomatous polyposis ในประวัติของผู้ป่วย ได้แก่:
- กลุ่มอาการ Hamartoma หลายแห่ง - กลุ่มอาการ Cowden ซึ่งมีการเกิด Hamartoma หลายแห่งที่มีต้นกำเนิดจาก ecto- ento- และ mesodermal โดยมีอาการ polyposis ในระบบทางเดินอาหารและอาการทางผิวหนังและเยื่อเมือก
- กลุ่มอาการ Peutz-Jeghers-Turen (มีลักษณะเฉพาะคือมีการพัฒนาของเนื้องอกชนิด hamartomatous ที่ไม่ร้ายแรงในทางเดินอาหาร)
- โรคโพรตีอุส;
- โรค Weil - เนื้องอกลำไส้ใหญ่ชนิดไม่ร้ายแรง;
- กลุ่มอาการ Bannayan-Riley-Ruvalcaba ซึ่งเช่นเดียวกับกลุ่มอาการ Cowden ทำให้เกิด hamartoma (hamartomatous polyps) หลายแห่งในลำไส้
- กลุ่มอาการคาร์นีย์-สตราตาคิสและกลุ่มอาการคาร์นีย์
นอกจากนี้ hamartomas ยังเกิดขึ้นในผู้ป่วยที่มีกลุ่มอาการ Watson ที่ถ่ายทอดทางพันธุกรรม และในกรณีที่เกิดเป็นครั้งคราวหรือเป็นกลุ่มอาการ Pallister-Hall แต่กำเนิดร่วมกับ hamartoma จากไฮโปทาลามัสและนิ้วหลายนิ้ว
กลไกการเกิดโรค
กลไกในการเพิ่มการแบ่งตัวของเนื้อเยื่อเจริญด้วยการก่อตัวของความผิดปกติคล้ายเนื้องอกในอวัยวะต่างๆ ได้รับการอธิบายโดยความผิดปกติของโครโมโซมและการกลายพันธุ์ของยีนที่อาจเกิดขึ้นได้เองตามธรรมชาติหรือถ่ายทอดทางพันธุกรรม
ในโรค Tuberous Sclerosis พบการกลายพันธุ์ในยีน TSC1 หรือ TSC2 ซึ่งเป็นยีนระงับเนื้องอกที่ป้องกันและยับยั้งการแบ่งตัวของเซลล์ที่มากเกินไป เช่น การเจริญเติบโตและการแบ่งตัวของเซลล์ที่รวดเร็วเกินไปหรือควบคุมไม่ได้ และในโรค neurofibromatosis ชนิดที่ 1 และกลุ่มอาการ Watson พบการกลายพันธุ์ของยีน NF1 ซึ่งเป็นยีนระงับเนื้องอกในไมโตคอนเดรีย
ในกลุ่มอาการเนื้องอก hamartoma ซึ่งรวมถึงกลุ่มอาการ Cowden, Protea, Bannayan-Riley-Ruvalcaba และโรคโพลีโพซิสในเด็ก การเกิดโรคจะเกี่ยวข้องกับการกลายพันธุ์ของยีน PTEN ซึ่งเข้ารหัสเอนไซม์ที่เกี่ยวข้องกับการควบคุมการแพร่กระจายและถือเป็นยีนระงับเนื้องอก
การกลายพันธุ์ในยีน STK11 ซึ่งเข้ารหัสโครงสร้างและหน้าที่ของเอนไซม์เซอรีนทรานส์เมมเบรนชนิดหนึ่ง ซึ่งลดความสามารถในการยับยั้งการแบ่งตัวของเซลล์ ทำให้เกิดกลุ่มอาการ Peutz-Jeghers-Turen ซึ่งทำให้เกิดติ่งเนื้อในลำไส้และรอยโรคบนผิวหนังที่มีเม็ดสี การกลายพันธุ์ในยีน GLI3 ซึ่งเป็นปัจจัยการถอดรหัสที่เกี่ยวข้องกับการสร้างเนื้อเยื่อภายในมดลูก ได้รับการระบุในกลุ่มอาการ Pallister-Hall
ดังนั้นการเจริญเติบโตของเซลล์ที่ไม่สามารถควบคุมได้ซึ่งเกิดจากการกลายพันธุ์ของยีนทำให้เกิดการสร้างฮามาร์โตมา
อาการ ของฮามาร์โทมา
เนื้องอกแฮมารโตมาแบ่งออกเป็นประเภทต่างๆ ขึ้นอยู่กับตำแหน่งของเนื้องอก และแต่ละชนิดจะมีโครงสร้างและอาการเฉพาะของตัวเอง
เนื้องอกของปอด
เนื้องอกในปอดสามารถเกิดขึ้นได้ในทุกกลีบปอดและส่วนปลายของปอด และประกอบด้วยเนื้อเยื่อปกติที่มีอยู่ในปอด ได้แก่ เนื้อเยื่อไขมัน เนื้อเยื่อบุผิว เนื้อเยื่อเส้นใย และเนื้อเยื่อกระดูกอ่อน ใน 80% ของกรณี ส่วนประกอบของคอนโดรด (เซลล์กระดูกอ่อนใส) มักประกอบด้วยเซลล์ไขมัน เซลล์เนื้อเยื่อไขมันและเซลล์เยื่อบุทางเดินหายใจ [ 3 ]
ชื่อเดิม ได้แก่ chondroid hamartoma, mesenchymoma, chondromatous hamartoma หรือ hamartochondroma ปัจจุบัน WHO ยังไม่แนะนำให้ใช้
ในทางกลับกัน ซีสต์ฮามาร์โตมาของเนื้อเยื่อเกี่ยวพันในปอดนั้นพบได้น้อยกว่า และมักเกี่ยวข้องกับกลุ่มอาการโคเดนในผู้ป่วยส่วนใหญ่
โรคปอดบวมอาจไม่แสดงอาการ แต่สามารถทำให้เกิดอาการต่างๆ เช่น ไอเรื้อรัง (มักมีอาการไอเป็นเลือด) หายใจมีเสียงหวีด และหายใจลำบาก [ 4 ]
แฮมมาโตมาของหัวใจ
เนื้องอกหัวใจหลักชนิดไม่ร้ายแรงในผู้ใหญ่ ได้แก่ myocyte hamartoma ที่โตเต็มที่ และในทารกและเด็กที่เป็นโรค Tuberous Sclerosis หรือ Rhabdomyoma นั่นคือ hamartoma ของกล้ามเนื้อหัวใจที่โพรงหัวใจหรือผนังกั้นระหว่างโพรงหัวใจ [ 5 ]
แฮมมาร์โตมาของเซลล์กล้ามเนื้อหัวใจที่โตเต็มที่จะเกิดขึ้นที่ผนังห้องล่างของหัวใจ (และพบได้น้อยที่ห้องบน) และอาจปรากฏเป็นรอยโรคหลายจุดที่มีความหนาแน่นและเกี่ยวข้องอย่างใกล้ชิดกับกล้ามเนื้อหัวใจที่อยู่ด้านล่าง เนื้องอกอาจทำให้เกิดอาการของภาวะหัวใจล้มเหลว ได้แก่ อาการเจ็บหน้าอก ใจสั่นและหัวใจเต้นผิดจังหวะ เสียงหัวใจเต้นผิดปกติ อาการบวมน้ำ หายใจลำบาก และตัวเขียว
เนื้องอกกล้ามเนื้อหัวใจซึ่งส่วนใหญ่ได้รับการวินิจฉัยภายในปีแรกของชีวิต ประกอบด้วยเนื้อเยื่อของกล้ามเนื้อหัวใจที่ก่อตัวจากไมโอบลาสต์ของตัวอ่อน และมีลักษณะเป็นก้อนเนื้อแข็งๆ ที่ไม่มีแคปซูล
โดยทั่วไป hamartomas เหล่านี้จะปรากฏขึ้นโดยไม่มีอาการ และจะค่อยๆ หายไปเองก่อนอายุ 4 ปี
ผู้เชี่ยวชาญบางคนยังถือว่ารอยโรคแบบ Hamartoma เกี่ยวข้องกับ Carney complex myxoma ของหัวใจ ด้วย [ 6 ]
แกมาร์โตมาของทางเดินอาหาร
Gastric hamartoma คือก้อนเนื้อเนื้อเยื่อเกี่ยวพันที่มีรูปร่างเป็นเยื่อบุผิวที่ขยายใหญ่ขึ้นในกระเพาะอาหาร โพลิป Peutz-Jeghers และ Myoepithelial hamartoma ที่หายาก ซึ่งมีมัดกล้ามเนื้อเรียบที่ขยายใหญ่ขึ้น ชื่ออื่นๆ ของ hamartoma นี้ ได้แก่ myoglandular hamartoma, adenomyomatous hamartoma และ gastric adenomyoma อาการทางคลินิกทั่วไป ได้แก่ อาการอาหารไม่ย่อย ปวดท้องน้อย และเลือดออกในทางเดินอาหารส่วนบน [ 7 ], [ 8 ]
ข้อมูลเพิ่มเติมในเอกสาร - โรคกระเพาะมีติ่ง
เนื้องอกในลำไส้คือเนื้องอกในลำไส้ใหญ่ ที่มีเนื้องอกในลำไส้ใหญ่หรือ เนื้องอกในท่อ เนื้องอกนี้ได้รับการวินิจฉัยว่าเป็นเนื้องอกต่อมน้ำเหลืองหรือเนื้องอกในท่อ เมื่อเนื้องอกนี้ไปอยู่ที่ต่อมบรุนเนอร์ของลำไส้เล็กส่วนต้น อาการจะแสดงออกมาด้วยอาการปวดบริเวณเหนือกระเพาะอาหาร คลื่นไส้ อาเจียน และท้องอืด (ซึ่งบ่งชี้ถึงการอุดตันของลำไส้) และหากเนื้องอกมีขนาดใหญ่ อาจมีเลือดออกในทางเดินอาหาร ในกรณีของเนื้องอกในกล้ามเนื้อเรียบของลำไส้เล็กส่วนปลาย ผู้ป่วยจะบ่นว่าปวดท้อง น้ำหนักลด และเกิดภาวะโลหิตจางเรื้อรัง [ 9 ], [ 10 ]
อ่านเพิ่มเติม - โพลิปทวารหนัก
Retrorectal hamartoma คือซีสต์ hamartoma หรือซีสต์หลายช่องในช่อง retrorectal (เนื้อเยื่อเกี่ยวพันหลวมๆ ระหว่างทวารหนักและพังผืดของทวารหนัก) ที่มักเกิดขึ้นในผู้หญิงวัยกลางคน มีลักษณะเป็นซีสต์ที่นูนออกมาจากผนังด้านหลังของทวารหนัก ซึ่งบุด้วยเยื่อบุผิวและมีเส้นใยกล้ามเนื้อเรียบเรียงกันอย่างไม่เป็นระเบียบ Hamartoma นี้มีอาการปวดท้องน้อยและท้องผูกเป็นประจำ [ 11 ], [ 12 ]
เนื้องอกของตับและม้าม
แฮมมาร์โตมาท่อน้ำดีหลายท่อในตับเป็นแฮมมาร์โตมาของท่อน้ำดีในตับระหว่างตุ๊กตาซึ่งเกี่ยวข้องกับความผิดปกติของการพัฒนาในระยะตัวอ่อน แฮมมาร์โตมา (ท่อเดียวหรือหลายท่อ) นี้ประกอบด้วยกลุ่มท่อน้ำดีที่ขยายตัวแบบไร้ทิศทางและเนื้อเยื่อเกี่ยวพันที่เป็นเส้นใยคอลลาเจน [ 13 ]
เนื้องอกท่อน้ำดีแบบ hamartomas มักไม่มีอาการและมักถูกค้นพบโดยบังเอิญ (ระหว่างการตรวจทางรังสีวิทยาหรือการผ่าตัดเปิดหน้าท้อง) [ 14 ]
เนื้องอกชนิดไม่ร้ายแรงที่พบได้น้อยและมักตรวจพบโดยบังเอิญคือเนื้องอกแฮมมาร์โตมาของม้าม ซึ่งประกอบด้วยเนื้อเยื่อสีแดงของม้าม มีลักษณะเป็นก้อนเนื้อแข็งที่มีรูปร่างชัดเจนและสม่ำเสมอ ความผิดปกตินี้สามารถเกิดขึ้นได้ทีละก้อนหรือหลายก้อน เมื่อบีบเนื้อม้าม อาจรู้สึกไม่สบายและเจ็บปวดที่บริเวณใต้ชายโครงด้านซ้าย [ 15 ], [ 16 ]
เนื้องอกของไต
เนื้องอกแฮมมาร์โตมาของไตที่พบได้บ่อยที่สุดได้รับการวินิจฉัยว่าเป็นเนื้องอกแองจิโอไมโอลิโปมาของไตเนื่องจากเนื้องอกชนิดนี้ไม่ร้ายแรง ประกอบด้วยเนื้อเยื่อไขมันที่โตเต็มที่ซึ่งมีเส้นใยกล้ามเนื้อเรียบฝังตัวและหลอดเลือด เนื้องอกชนิดนี้เกิดขึ้นในผู้ป่วยโรคสเคอโรซิสในร้อยละ 40-80 ขนาดของเนื้องอกแฮมมาร์โตมาที่เพิ่มขึ้น (มากกว่า 4-5 ซม.) จะทำให้มีอาการปวดและมีเลือดในปัสสาวะ [ 17 ], [ 18 ]
แฮมารโตมาบริเวณเต้านม
คำจำกัดความการวินิจฉัยโรคเต้านม hamartoma ที่องค์การอนามัยโลกยอมรับ ได้แก่ adenolipoma, chondrolipoma และ myoid hamartoma แม้ว่านักเต้านมวิทยาจะเรียก fibroadenolipoma บ่อยครั้ง เนื่องจากการก่อตัวของเนื้องอกประกอบด้วยเซลล์ของเนื้อเยื่อเกี่ยวพันบางๆ ต่อม และไขมันที่ห่อหุ้มอยู่ในแคปซูลเนื้อเยื่อเกี่ยวพันบางๆ ที่มีโครงร่างชัดเจน อาจสังเกตเห็นการสะสมของแคลเซียมเฉพาะจุดเมื่อมองเห็น ในกรณีนี้ จะไม่มีการแสดงอาการทางคลินิก [ 19 ], [ 20 ]
อ่านเพิ่มเติม - เนื้องอกเต้านม
เนื้องอกของสมอง
ผู้ป่วยโรค Tuberous Sclerosis หนึ่งในสามรายมีเนื้องอกในสมองในรูปแบบของเนื้องอกในสมองที่งอกออกมาหรือเนื้องอกในกลีบสมองต่างๆ (บริเวณขอบของเนื้อสมองสีเทาและสีขาว) หรือมีปุ่มใต้เยื่อบุโพรงสมองตามผนังโพรงสมอง เนื้องอกในสมองชนิด Astrocytic hamartoma ซึ่งเป็นเนื้องอกในสมองชนิดเซลล์ยักษ์ใต้เยื่อบุโพรงสมองที่มีการแตกของเยื่อหุ้มสมอง เซลล์ประสาทผิดปกติ และเซลล์เกลียขนาดใหญ่ของเนื้อสมอง (เซลล์แอสโตรไซต์) อาจก่อตัวขึ้นได้เช่นกัน อาการของเนื้องอกในสมองชนิด hamartoma ได้แก่ อาการชักและปัญญาอ่อนในเด็ก [ 21 ], [ 22 ]
ความผิดปกติที่พบได้น้อยซึ่งเกิดขึ้นระหว่างการสร้างตัวอ่อนและปรากฏให้เห็นตั้งแต่แรกเกิดคือ ไฮโปทาลามัสฮามาร์โตมา ซึ่งเป็นกลุ่มของเซลล์ประสาทเฮเทอโรโทปิกและเซลล์เกลีย เมื่อสมองของเด็กเติบโต เนื้องอกจะขยายใหญ่ขึ้นแต่จะไม่แพร่กระจายไปยังบริเวณสมองอื่น [ 23 ], [ 24 ]
หากมีการสร้างเนื้อเยื่อที่โตเกินในส่วนหน้าของไฮโปทาลามัส (tuber cinereum) ซึ่งเป็นบริเวณที่ต่อมใต้สมองเชื่อมติดอยู่ ความผิดปกติจะแสดงอาการของพัฒนาการทางเพศก่อนวัยอันควรของระบบประสาทส่วนกลาง (ก่อนอายุ 8-9 ปี) ดังนี้ ผื่นสิวปรากฏ ต่อมน้ำนมพัฒนาเร็วและมีประจำเดือนครั้งแรกเร็วในเด็กหญิง ขนเพชรและเสียงผิดปกติก่อนวัยในเด็กชาย
เมื่อ hamartomas ก่อตัวในส่วนหลังของไฮโปทาลามัส อาจมีความผิดปกติในกิจกรรมทางไฟฟ้าของสมอง โดยในวัยทารกจะแสดงออกด้วยอาการชัก และในระยะต่อมา (อายุ 4-7 ปี) อาจมีอาการลมบ้าหมูที่มีอาการชักแบบเฉพาะที่พร้อมกับหัวเราะอย่างกะทันหันหรือร้องไห้โดยไม่ได้ตั้งใจ อาการชักแบบอะโทนิกหรือแบบเกร็งกระตุก รวมถึงอาการชักแบบก้าวร้าว ความจำ และปัญหาทางสติปัญญา
ต่อมใต้สมอง Hamartoma คือเนื้องอกของต่อมใต้สมองชนิด ไม่ร้ายแรงที่เกิดขึ้นเป็นครั้ง คราว
ผู้ใหญ่วัยกลางคนที่เป็นโรค Cowden อาจมีก้อนเนื้อคล้ายเนื้องอกที่หายาก เรียกว่า hamartoma ของสมองน้อย ซึ่งวินิจฉัยว่าเป็น dysplastic cerebellar gangliocytoma หรือโรค Lhermitte-Duclos อาการอาจไม่ปรากฏหรือแสดงออกมาเป็นอาการปวดศีรษะ เวียนศีรษะ การประสานงานการเคลื่อนไหวบกพร่อง และอัมพาตของเส้นประสาทสมองแต่ละข้าง
ต่อมน้ำเหลืองโตแบบฮามาร์โตมา
เมื่อเซลล์ของกล้ามเนื้อเรียบและเนื้อเยื่อไขมัน รวมทั้งหลอดเลือดและเนื้อเยื่อเกี่ยวพันคอลลาเจนของต่อมน้ำเหลืองบริเวณขาหนีบ หลังเยื่อบุช่องท้อง ใต้ขากรรไกร และปากมดลูก เจริญเติบโตมากเกินไป จะเกิด angiomyomatous hamartoma ของต่อมน้ำเหลืองหรือ nodular angiomyomatous hamartoma โดยมีการทดแทนเนื้อของต่อมน้ำเหลืองบางส่วนหรือทั้งหมด [ 25 ], [ 26 ]
แฮมารโตมาของผิวหนัง
ในกรณีที่มีภาวะ tuberous sclerosis หรือ neurofibromatosis จะสังเกตเห็น hamartomas ต่างๆ ของผิวหนัง โดยส่วนใหญ่มักจะอยู่ในรูปแบบของจุดที่มีเม็ดสีลดลง จุดกาแฟและจุดนมเนื้องอกหลอดเลือด (ที่แก้ม คาง รอยพับระหว่างร่องแก้ม) จุด shagreen ในตำแหน่งต่างๆ (ซึ่งคือ เนวัสของเนื้อเยื่อเกี่ยวพัน) คราบพังผืดบนหน้าผาก หนังศีรษะ หรือคอ
การแสดงออกทางผิวหนังที่พบได้น้อยของโรค Tuberous Sclerosis (โดยเฉพาะในผู้ชาย) คือ Folliculocystic และ Collagen hamartoma ซึ่งมีลักษณะเด่นคือมีการสะสมของคอลลาเจนจำนวนมากในชั้นหนังแท้ มีพังผืดรอบรูขุมขนที่รวมกันเป็นวงกลม และมีซีสต์ใต้ผิวหนังรูปกรวยที่เต็มไปด้วยเคราติน ซึ่งพบได้ในการตรวจทางพยาธิวิทยา [ 27 ]
สำหรับ hamartomas ที่ประกอบด้วยเมลาโนไซต์ (เซลล์ที่สร้างเม็ดสีเมลานิน) ผู้เชี่ยวชาญส่วนใหญ่ยังหมายถึงเนื้องอกเมลาโนไซต์ ชนิดต่างๆ โดยเฉพาะเนวัสเมลาโนไซต์ที่เกิดแต่กำเนิดซึ่งเป็นความผิดปกติของการสร้างตัวอ่อน
เมื่อพิจารณาจากสาเหตุ hamartomas ที่ประกอบด้วยเนื้อเยื่อหลอดเลือด ถือเป็นhemangiomas ของผิวหนัง ด้วยเช่นกัน
ผู้ป่วยที่มีอาการ Peutz-Jeghers-Thuren จะมีเนื้องอกที่เรียกว่า hamartoma ในรูปแบบของเม็ดสีที่ไม่สม่ำเสมอบนผิวหนังและเยื่อเมือก - lentiginosis periorificialis
ในกรณีของเนื้องอกแฮมารโตมาชนิดเอ็กโตเดอร์มัล-เมโซเดอร์มัลเชิงเส้น (Hamartoma moniliformis) จะแสดงอาการผื่นเป็นตุ่มสีเนื้อเชิงเส้นที่ศีรษะ คอ และหน้าอกส่วนบน
และ sebocytic hamartoma คือ hamartoma ของต่อมไขมัน อ่านเพิ่มเติมในสิ่งพิมพ์ - sebaceous nevus
แฮมมาโตมาของดวงตา
รอยโรคม่านตาที่มีเม็ดสีแบบ hamartomatous ในโรคเนื้องอกเส้นประสาทชนิดที่ 1 และโรค Watson มีลักษณะเป็นกลุ่มของเซลล์เมลาโนไซต์แบบเดนไดรต์เป็นปุ่ม เรียกว่า iris hamartoma หรือ Lisch nodules รอยโรคดังกล่าวเป็นตุ่มใสรูปโดมสีเหลืองน้ำตาลที่ยื่นออกมาเหนือพื้นผิวของม่านตา (โดยปกติจะไม่ส่งผลต่อการมองเห็น)
และผู้ป่วยที่มีเนื้องอกหลอดเลือดชนิดไม่รุนแรงในช่องจมูกและเนื้องอกชนิดมีต่อมไขมันแบบทางพันธุกรรมมักเกิดเนื้องอกฮามาร์โตมาของจอประสาทตาและเยื่อบุผิวเรตินัลพิกเมนต์ร่วมกัน ซึ่งมีลักษณะเป็นจุดดำที่ส่วนกลาง (จุดรับภาพ) ของจอประสาทตา [ 28 ]
แฮมมาโตมาของจมูก
ผู้เชี่ยวชาญให้คำจำกัดความของ Nasal hamartoma ว่าเป็น Nasal chondromesenchymal hamartoma หรือNasal chondromaซึ่งเกิดจากการขยายพันธุ์ของเยื่อบุผิวทางเดินหายใจ ต่อมใต้เยื่อเมือก และเนื้อเยื่อเกี่ยวพันของกระดูกอ่อน อาการทางคลินิกขึ้นอยู่กับขนาดและตำแหน่งของรอยโรค ได้แก่ อาการคัดจมูก หายใจลำบากและให้นมลูกได้ยาก น้ำมูกใสไหล และเลือดออกทางจมูก Hamartoma อาจเติบโตไปพร้อมกับเด็กและแพร่กระจายไปยังเบ้าตา ส่งผลให้ลูกตาเคลื่อนไปข้างหน้าหรือข้างหลัง ตาเหล่ หรือความผิดปกติของกล้ามเนื้อตา [ 29 ]
แฮมารโตมาในเด็ก
โรค hamartomatous ของอวัยวะต่างๆ และโครงสร้างทางกายวิภาคที่กล่าวมาทั้งหมดเกิดขึ้นในเด็กที่มีอาการดังต่อไปนี้
ทารกแรกเกิดมีเนื้องอกเนื้อเยื่อเกี่ยวพันของผนังทรวงอกหรือเนื้องอกเนื้อเยื่อเกี่ยวพันของกระดูกอ่อนของซี่โครง ซึ่งเป็นก้อนเนื้อแข็งที่เคลื่อนไหวไม่ได้ซึ่งเกิดจากการเติบโตมากเกินไปของโครงกระดูกปกติที่มีองค์ประกอบของกระดูกอ่อน หลอดเลือด และเนื้อเยื่อเกี่ยวพัน เนื้องอกเนื้อเยื่อเกี่ยวพันนี้อาจทำให้เกิดภาวะระบบทางเดินหายใจล้มเหลวและเกิดกลุ่มอาการหายใจลำบาก เนื้องอกเนื้อเยื่อเกี่ยวพันของตับเป็นเนื้องอกตับชนิดไม่ร้ายแรง ที่พบบ่อยเป็นอันดับสอง ในเด็ก การก่อตัวคล้ายเนื้องอกนี้ (มักเกิดขึ้นที่กลีบขวาของอวัยวะ) ประกอบด้วยเซลล์ของเนื้อเยื่อเกี่ยวพันของเนื้อเยื่อเกี่ยวพัน เซลล์ตับ และเซลล์เยื่อบุผิวของเยื่อบุท่อน้ำดี ภาพทางคลินิกได้แก่ ก้อนเนื้อที่สัมผัสได้ในช่องท้อง เบื่ออาหารและน้ำหนักลด และในกรณีที่มีขนาดใหญ่มาก (มากถึง 10 ซม. ขึ้นไป) เนื้องอกจะปกคลุมท่อน้ำดีนอกตับและหลอดเลือดดำใหญ่ด้านล่าง ทำให้เกิดอาการตัวเหลืองและอาการบวมน้ำที่บริเวณขาส่วนล่าง
Hamartoma คือเนื้องอกเนโฟมาที่มีมาแต่กำเนิด (เกิดในทารก 1 ใน 200,000 ราย) ซึ่งอาจทำให้ทารกแรกเกิดมีอาการท้องอืดและคลำพบก้อนเนื้อหนาแน่นที่ช่องท้องด้านขวาบน ทารกอาจหายใจเร็วและตื้นด้วย
ความผิดปกติแต่กำเนิดที่หายาก ได้แก่ เนื้องอกเส้นใยในวัยทารก ซึ่งเกิดขึ้นในเด็กในช่วงสองปีแรกของชีวิต และแสดงอาการเป็นก้อนเนื้อไม่เจ็บปวดในเนื้อเยื่อใต้ผิวหนังของรักแร้ คอ ไหล่และปลายแขน หลังและหน้าอก ต้นขา เท้า และอวัยวะเพศภายนอก
เนื้องอกต่อมเหงื่อเอคครินชนิดแอนจิโอมาในเด็กอาจเกิดขึ้นตั้งแต่แรกเกิดหรือปรากฏให้เห็นในวัยเด็ก เนื้องอกชนิดไม่ร้ายแรงนี้มักมีลักษณะเป็นก้อนเนื้อสีน้ำเงินหรือน้ำตาลและ/หรือเป็นแผ่น ซึ่งเกิดจากการเพิ่มจำนวนเนื้อเยื่อต่อมเหงื่อเอคครินและเส้นเลือดฝอยในชั้นกลางและชั้นลึกของหนังแท้ เนื้องอกต่อมเหงื่อชนิดนี้อาจทำให้เกิดภาวะเหงื่อออกมากเกินไปในบริเวณเฉพาะที่และผมยาวขึ้น
ภาวะแทรกซ้อนและผลกระทบ
โดยทั่วไปแล้ว เป็นที่ยอมรับกันว่า hamartomas มักไม่กลับมาเป็นซ้ำหรือกลายเป็นเนื้องอกร้าย โดยมักมีอาการเพียงเล็กน้อยหรือไม่มีอาการเลย และบางครั้งอาจหายไปเมื่อเวลาผ่านไป แต่ในกรณีที่รุนแรงกว่านั้นและขึ้นอยู่กับตำแหน่งที่เกิด ความผิดปกติเหล่านี้อาจก่อให้เกิดภาวะแทรกซ้อนและผลที่ตามมาที่ร้ายแรงได้
ประการแรก hamartoma อาจเติบโตจนมีขนาดใหญ่จนเริ่มกดทับเนื้อเยื่อและอวัยวะโดยรอบ ทำให้การทำงานของอวัยวะเหล่านั้นผิดปกติ
ภาวะหัวใจหยุดเต้นเฉียบพลันในเด็กอาจส่งผลให้เกิดความผิดปกติของจังหวะการเต้นของหัวใจอย่างต่อเนื่อง ความผิดปกติของลิ้นหัวใจ และการไหลเวียนเลือดภายในหัวใจบกพร่อง ซึ่งส่งผลให้เกิดภาวะหัวใจล้มเหลวตามมา
ภาวะแทรกซ้อนของการเกิด hamartomatous polyps ในทางเดินอาหาร ได้แก่ เลือดออกในทางเดินอาหาร การอุดตัน และภาวะลำไส้สอดเข้าไป (ซึ่งอาจถึงแก่ชีวิตได้) และ hamartoma ขนาดใหญ่ของไตอาจทำให้ไตแตกได้
ภาวะ แฮมารโตมาในสมองอาจทำให้เกิดภาวะโพรงสมองอุดตันได้
ในเนื้องอกต่อมใต้สมองและไฮโปทาลามัส การผลิตฮอร์โมนการเจริญเติบโต (ฮอร์โมนการเจริญเติบโต) อาจบกพร่อง ส่งผลให้เกิดภาวะต่อมใต้สมองทำงานน้อย (hypopituitarism) ในเด็กเนื้องอกต่อมใต้สมองและไฮโปทาลามัสในเด็กอาจทำให้เกิดโรคลมบ้าหมูที่ดื้อยาได้เช่นกัน
ภาวะแทรกซ้อนของเยื่อบุผิวเรตินัลพิกเมนต์ฮามาร์โตมาเต็มไปด้วยปัญหาด้านจอประสาทตาและ/หรือเส้นประสาทตาทำงานผิดปกติ อาการบวมของจุดรับภาพ การสร้างหลอดเลือดใหม่ในโคโรอิด และจอประสาทตาหลุดลอก
การวินิจฉัย ของฮามาร์โทมา
ส่วนสำคัญอย่างหนึ่งในการวินิจฉัย hamartoma และกลุ่มอาการที่เกี่ยวข้องคือการรวบรวมประวัติทางการแพทย์รวมทั้งประวัติครอบครัว
การทดสอบในห้องปฏิบัติการ ได้แก่ การตรวจเลือดทั่วไป การตรวจอิเล็กโทรไลต์ในซีรั่ม การตรวจระดับลิมโฟไซต์ ระดับแคลเซียม โพแทสเซียม ฟอสเฟต และยูเรีย และการทดสอบการทำงานของตับ หากเป็นไปได้ จะทำการตรวจชิ้นเนื้อด้วยเข็มขนาดเล็กเพื่อดูดก้อนเนื้อ เนื่องจากการตรวจทางจุลพยาธิวิทยามีความสำคัญอย่างยิ่งในการวินิจฉัยและการเลือกวิธีการรักษา
การวินิจฉัยด้วยเครื่องมือช่วยให้มองเห็นการก่อตัวของเนื้องอกแบบ hamartomatous และระบุตำแหน่งที่แน่นอนของเนื้องอกได้ โดยจะใช้การเอกซเรย์ การถ่ายภาพหลอดเลือด คลื่นไฟฟ้าสมอง (EEG) อัลตราซาวนด์ (โซโนกราฟี) CT (เอกซเรย์คอมพิวเตอร์) PET (เอกซเรย์ปล่อยโพซิตรอน) และ MRI (การถ่ายภาพด้วยคลื่นแม่เหล็กไฟฟ้า)
การวินิจฉัยที่แตกต่างกัน
การวินิจฉัยแยกโรคมีความสำคัญมากสำหรับก้อนเนื้อที่ผิดปกติ ดังนั้น เนื้องอกวัณโรคและเนื้องอกฮามาร์โตมาจึงถูกแยกออกจากเนื้องอกฮามาร์โตมาในปอดและมะเร็งปอดขั้นต้น เนื้องอกคาร์ซินอยด์จากหลอดลม และโรคที่แพร่กระจาย ควรแยกเนื้องอกฮามาร์โตมาในสมองออกจากเนื้องอกคอหอยและเนื้องอกในสมองไฮโปทาลามัส-ไคแอสมา และการวินิจฉัยแยกโรคฮามาร์โตมาว่าเป็นเนื้องอกเนฟโรมาเมโสบลาสติกแต่กำเนิด ได้แก่ เนื้องอกวิลม์ส (เนื้องอกเนฟโรบลาสโตมาชนิดร้ายแรง) เนื้อเยื่อเกี่ยวพันเซลล์ใสของไต และเนื้องอกไตที่แข็งตัวในทารก
ใครจะติดต่อได้บ้าง?
การรักษา ของฮามาร์โทมา
หากฮามาร์โตมาไม่มีอาการและตรวจพบโดยบังเอิญ ก็ไม่จำเป็นต้องรักษา แต่จำเป็นต้องติดตาม "พฤติกรรม" ของฮามาร์โตมาและสภาพของผู้ป่วย ในกรณีอื่นๆ การบำบัดมีจุดมุ่งหมายเพื่อลดความรุนแรงของอาการและป้องกันภาวะแทรกซ้อน ตัวอย่างเช่น ในฮามาร์โตมาของไฮโปทาลามัสที่มีอาการของการเข้าสู่วัยรุ่นก่อนวัยอันควร แพทย์จะสั่งจ่ายยาบางชนิดที่ยับยั้งการปล่อยฮอร์โมนบางชนิด ยารักษาโรคหัวใจใช้เพื่อรักษาอาการของโรคหัวใจล้มเหลวในผู้ป่วยฮามาร์โตมาของหัวใจ
การผ่าตัดเอาเนื้องอกแฮมมาร์โตมาออกมีข้อบ่งชี้เพื่อยืนยันการวินิจฉัย และในกรณีที่มีอาการรุนแรงที่ไม่สามารถแก้ไขได้ทางการแพทย์
ตัวอย่างเช่น เนื้องอกของปอดอาจถูกตัดออกโดยการตัดลิ่ม และในรายที่รุนแรง อาจตัดปอดออกบางส่วน (การตัดกลีบปอดออก) เนื้องอกของเต้านมอาจถูกตัดออกได้เช่นกัน และหากเนื้องอกมีขนาดใหญ่ อาจต้องตัดเต้านมออกบางส่วนหรือทั้งหมด
การทำลายเนื้อเยื่อด้วยคลื่นวิทยุแบบสเตอริโอแทกติกหรือการขจัดเนื้อเยื่อด้วยเลเซอร์สามารถนำไปใช้เพื่อกำจัดเนื้องอกที่มีต่อมไขมันโตได้ นอกจากนี้ ยังใช้การผ่าตัดด้วยรังสีแกมมาที่มีความเข้มข้นสูง เช่น การผ่าตัดเนื้องอกที่มีต่อมไขมันโตในไฮโปทาลามัสหรือเนื้องอกที่มีต่อมไขมันโตในแอสโตรไซต์
การป้องกัน
วิธีเดียวที่จะป้องกันการเกิด hamartomas ได้คือการตรวจคัดกรองทางพันธุกรรมของพ่อแม่ในอนาคตของเด็ก
พยากรณ์
การพยากรณ์โรคโดยรวมของความผิดปกติแต่กำเนิดนี้ขึ้นอยู่กับตำแหน่งและขนาดของเนื้องอก รวมไปถึงภาวะแทรกซ้อนและสุขภาพทั่วไปของผู้ป่วย