ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
เนื้องอกของเซลล์ประสาทในเด็ก: สาเหตุ การวินิจฉัย การรักษา
ตรวจสอบล่าสุด: 04.07.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
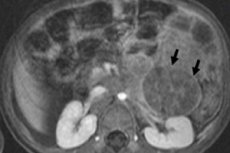
ในสาขาเนื้องอกวิทยาในเด็ก เนื้องอกนอกกะโหลกศีรษะที่พบบ่อยที่สุดชนิดหนึ่งคือเนื้องอกของนิวโรบลาสโตมาในเด็ก ซึ่งเป็นเนื้องอกร้ายของเอ็มบริโอในกลุ่มนิวโรบลาสต์ของนิวรัลเครสต์ หรือก็คือเซลล์ประสาทเอ็มบริโอ (ยังไม่โตเต็มที่) ของระบบประสาทซิมพาเทติก
ระบาดวิทยา
ตามสถิติของ International Neuroblastoma Risk Group (INRG) โรคมะเร็งต่อมหมวกไตคิดเป็นประมาณ 8% ของโรคเนื้องอกในเด็กทั้งหมดทั่วโลก และมีอุบัติการณ์สูงเป็นอันดับสาม รองจากโรคมะเร็งเม็ดเลือดขาวและเนื้องอกในสมอง
จากข้อมูลอื่นๆ พบว่าเนื้องอกของเซลล์ประสาทคิดเป็นประมาณ 28% ของมะเร็งทั้งหมดในทารก มากกว่าหนึ่งในสามของผู้ป่วยเนื้องอกของเซลล์ประสาทได้รับการวินิจฉัยในเด็กอายุต่ำกว่า 1 ปี อายุเฉลี่ยของการวินิจฉัยคือ 19-22 เดือน ผู้ป่วยที่ได้รับการวินิจฉัยมากกว่า 90% เกิดขึ้นในเด็กอายุ 2-5 ปี (โดยส่วนใหญ่เป็นเด็กชาย) อุบัติการณ์สูงสุดพบในเด็กอายุ 2-3 ปี และผู้ป่วยในเด็กอายุมากกว่า 5 ปีคิดเป็นน้อยกว่า 10%
สาเหตุ เนื้องอกของระบบประสาท
ในการศึกษาสาเหตุของเนื้องอกของเซลล์ประสาท นักวิจัยสรุปได้ว่าเนื้องอกนี้ในเด็กเกิดจากการกลายพันธุ์ทางพันธุกรรมที่เกิดขึ้นเป็นระยะๆ ระหว่างการสร้างตัวอ่อนหรือการพัฒนาในระยะหลังคลอด แต่สาเหตุของการเปลี่ยนแปลงยีนเหล่านี้ยังไม่เป็นที่ทราบแน่ชัด เนื่องจากยังไม่มีการระบุอิทธิพลของปัจจัยแวดล้อมที่ก่อให้เกิดความพิการแต่กำเนิด
เนื้องอกเหล่านี้สามารถเกิดขึ้นได้ทุกที่ รวมถึงช่องกลางทรวงอก คอ ช่องท้อง ต่อมหมวกไต ไต กระดูกสันหลัง และกระดูกเชิงกราน
ในบางกรณี เนื้องอกของเซลล์ประสาทในทารกอาจเกี่ยวข้องกับการกลายพันธุ์ที่ถ่ายทอดทางพันธุกรรม โดยเฉพาะการกลายพันธุ์ในยีนของโปรตีนเยื่อหุ้มเซลล์ CD246 บนโครโมโซม 2 ซึ่งก็คือเอนไซม์ไทโรซีนไคเนส ALK ที่ควบคุมการสื่อสารระหว่างเซลล์และมีบทบาทสำคัญในการทำงานของระบบประสาท ในยีนของโปรตีน PHOX2B (บนโครโมโซม 4) ซึ่งเกี่ยวข้องกับการเจริญเติบโตของเซลล์ประสาท
นอกจากนี้ Neuroblastoma ยังอาจเกี่ยวข้องกับneurofibromatosis ชนิดที่ 1 ในเด็ก,กลุ่มอาการ Beckwith-Wiedemannและภาวะน้ำตาลในเลือดต่ำที่มีอินซูลินมากเกินไป (nesidioblastosis pancreatitis)
ปัจจัยเสี่ยง
ปัจจุบัน กรรมพันธุ์ได้รับการยอมรับว่าเป็นปัจจัยเสี่ยงต่อการเกิดเนื้องอกของเซลล์ประสาทในเด็ก เนื่องจากมีเนื้องอกชนิดนี้ในประวัติครอบครัว รวมถึงความผิดปกติแต่กำเนิดที่เกี่ยวข้องกับการกลายพันธุ์ของยีนในระหว่างการพัฒนาของทารกในครรภ์ โดยเฉพาะอย่างยิ่งในกรณีของการพัฒนาของเนื้องอกหลายชนิดในอวัยวะต่างๆ
นักวิจัยยังไม่ได้ระบุปัจจัยภายนอกใดๆ ที่เพิ่มความเสี่ยงของเนื้องอกนี้
กลไกการเกิดโรค
กลไกการพัฒนาของเนื้องอกของเซลล์ประสาทเกิดจากการรบกวนการแบ่งตัวและการเจริญเติบโตของเซลล์ยอดประสาท ซึ่งเป็นเซลล์สองข้างที่เกิดขึ้นที่ขอบของท่อประสาทจากชั้นเซลล์สืบพันธุ์นอกชั้นผิวหนังของตัวอ่อนมนุษย์ เซลล์เหล่านี้จะอพยพ (เคลื่อนที่) และแบ่งตัวเป็นเซลล์หลายประเภท ได้แก่ เซลล์ประสาทรับความรู้สึกและเซลล์ประสาทอัตโนมัติ เซลล์ประสาทต่อมไร้ท่อและเซลล์ของต่อมหมวกไต เซลล์ของกระดูกอ่อนและกระดูกกะโหลกศีรษะ รวมถึงเซลล์เม็ดสี
ในเนื้องอกของเซลล์ประสาท เซลล์ประสาทที่อพยพมาจะไม่โตเต็มที่ แต่จะเติบโตและแบ่งตัวต่อไป จนกลายเป็นเนื้องอก และพยาธิสภาพของการก่อตัวของเนื้องอกเกี่ยวข้องกับการกลายพันธุ์ของยีนดังต่อไปนี้:
- โดยมีการจำลองส่วนหนึ่งของลำดับโครโมโซมหรือการจำลองส่วนของยีน LMO1 บนโครโมโซม 11 ซึ่งเข้ารหัสโปรตีน RBTN1 ในเซลล์สันประสาทของตัวอ่อน
- โดยมีการเปลี่ยนแปลงในจำนวนสำเนาของยีน NBPF10 บนโครโมโซม 1q21.1 ซึ่งเข้ารหัสโปรตีน DUF1220 ซึ่งควบคุมการแพร่กระจายของเซลล์ต้นกำเนิดของระบบประสาทในมนุษย์ ความผิดปกติเหล่านี้ส่งผลให้โครโมโซมนี้ซ้ำซ้อนหรือหายไป ซึ่งก็คือไม่มีส่วนหนึ่งของดีเอ็นเอ
- โดยมีการเปลี่ยนแปลงในยีนยับยั้งเนื้องอก ATRX (บนโครโมโซม Xq21.1)
- โดยมีการปรากฏของสำเนาเพิ่มเติม (การขยาย) ของยีนปัจจัยการถอดรหัส N-Myc บนโครโมโซม 2 ซึ่งเข้ารหัสสำหรับปัจจัยการถอดรหัสตัวหนึ่ง (โปรตีนที่จับกับ DNA) ที่ควบคุมกิจกรรมของยีนอื่น ๆ และควบคุมการแพร่กระจายของเซลล์ตั้งต้นในระหว่างการสร้างโปรตีนสำหรับการสร้างเนื้อเยื่อและอวัยวะของทารกในครรภ์ การขยายตัวของยีนนี้จะเปลี่ยนให้เป็นออนโคยีนซึ่งกระตุ้นให้เกิดการหยุดชะงักของวงจรเซลล์ การแพร่กระจายของเซลล์เพิ่มขึ้น และการก่อตัวของเนื้องอก
อาการ เนื้องอกของระบบประสาท
สัญญาณแรกของโรคมะเร็งต่อมหมวกไตนั้นไม่จำเพาะเจาะจง และอาจมีอาการเบื่ออาหาร (และน้ำหนักลด) อ่อนเพลียเมื่อกินอาหาร มีไข้ และปวดข้อ
อาการทางคลินิกขึ้นอยู่กับตำแหน่งของเนื้องอกหลักและการมีการแพร่กระจาย (ซึ่งเกิดขึ้นใน 60-73% ของกรณี)
บ่อยครั้ง เนื้องอกของต่อมหมวกไตชนิดปฐมภูมิมักเกิดขึ้นที่ต่อมหมวกไตส่วนใน ซึ่งมีต้นกำเนิดคล้ายกับเซลล์ประสาท ในเด็กอายุน้อยกว่า 1 ปี เนื้องอกของต่อมหมวกไตจะได้รับการวินิจฉัยใน 35-40% ของกรณี อาการของโรคนี้ได้แก่ ปวดท้อง มีไข้ น้ำหนักลด ปวดกระดูก โลหิตจาง หรือกลุ่มอาการเปปเปอร์ (Pepper's syndrome) ซึ่งได้แก่ ความเสียหายของตับแบบกระจายร่วมกับตับโตรุนแรงและกลุ่มอาการหายใจลำบาก
เนื้องอกของเนื้อเยื่อเกี่ยวพันรอบเยื่อบุช่องท้องหรือเนื้องอกของเนื้อเยื่อเกี่ยวพันรอบเยื่อบุช่องท้องในเด็ก เมื่อโตขึ้น เนื้องอกจะเริ่มกดทับกระเพาะปัสสาวะหรือลำไส้ ทำให้เกิดปัญหาในการปัสสาวะหรืออุจจาระ ขาบวม (ในเด็กผู้ชาย เนื้องอกจะบวมที่ถุงอัณฑะ)
เนื้องอกของช่องกลางทรวงอกในเด็ก (เนื้องอกของช่องกลางทรวงอก) มักกดทับ vena cava ส่วนบน ซึ่งอาจทำให้ใบหน้า คอ แขน และหน้าอกส่วนบนบวม (ผิวหนังเปลี่ยนเป็นสีแดงอมน้ำเงินและมีปุ่มใต้ผิวหนัง) อาจมีอาการไอและมีเสียงหวีด หายใจลำบาก หรือกลืนลำบาก ต่อมน้ำเหลืองโตที่คอ เหนือกระดูกไหปลาร้า และรักแร้
การแพร่กระจายของเซลล์เนื้องอกไปสู่ไขกระดูกทำให้เกิดภาวะโลหิตจาง เกล็ดเลือดต่ำ และเม็ดเลือดขาวต่ำ มีแนวโน้มที่จะมีเลือดออก
และหากเกิดการแพร่กระจายไปยังบริเวณรอบดวงตา อาจทำให้เกิดรอยคล้ำหรือรอยฟกช้ำรอบดวงตาได้ นอกจากนี้ เนื้องอกดังกล่าวยังอาจทำให้เกิดอาการปวดศีรษะ เวียนศีรษะ ลูกตาโปน และปลายประสาทถูกกดทับ ทำให้หนังตาตก (ptosis) และรูม่านตาเล็กลง (miosis) เนื่องมาจากการกดทับ
เนื้องอกของกระเพาะหรือเนื้องอกของกระเพาะในเด็กทำให้เกิดการสร้างเนื้อเยื่อที่คลำได้ในช่องท้อง ท้องอืด เบื่ออาหาร ท้องผูก และความดันโลหิตสูง เนื้องอกที่กดทับไขสันหลังหรือรากประสาทอาจทำให้เกิดอาการชาและอ่อนแรงของแขนขา ไม่สามารถยืน คลาน หรือเดินได้ หากกระดูกได้รับผลกระทบ อาจทำให้เกิดอาการปวดกระดูก
ในกรณีของเนื้องอกระยะที่ 3-4 ในช่องท้องซึ่งต่อมน้ำเหลืองได้รับความเสียหาย เซลล์เนื้องอกสามารถแทรกเข้าไปในเนื้อไตได้ และอาจทำให้เกิดเนื้องอกของต่อมหมวกไตในเด็กได้ ส่งผลให้การทำงานของไตหยุดชะงัก
ขั้นตอน
- เนื้องอกของระบบประสาทระยะที่ 1 เป็นเนื้องอกหลักที่เกิดขึ้นในบริเวณใดบริเวณหนึ่งของร่างกาย ต่อมน้ำเหลืองทั้งสองข้างไม่ได้รับผลกระทบ
- เนื้องอกของเซลล์ประสาทระยะที่ 2 ในระยะที่ 2A เนื้องอกหลักจะอยู่ในจุดเดียวแต่มีขนาดใหญ่ ต่อมน้ำเหลืองทั้งสองข้างไม่ได้รับผลกระทบ ในระยะที่ 2B ต่อมน้ำเหลืองที่ด้านข้างของร่างกายที่เนื้องอกตั้งอยู่จะตรวจพบว่ามีการแพร่กระจาย
- เนื้องอกของระบบประสาทระยะที่ 3: เนื้องอกหลักลุกลามข้ามไขสันหลังหรือแนวกลางลำตัว พบการแพร่กระจายข้างเดียวหรือสองข้างที่ต่อมน้ำเหลือง
- เนื้องอกของเซลล์ประสาทระยะที่ 4: เนื้องอกได้แพร่กระจายไปยังต่อมน้ำเหลืองที่อยู่ไกลออกไป ไขกระดูก กระดูก ตับ หรืออวัยวะอื่น และระยะที่ 4S ตรวจพบในเด็กอายุน้อยกว่า 1 ปีที่มีเนื้องอกหลักอยู่เฉพาะที่ โดยมีการแพร่กระจายไปที่ผิวหนัง ตับ หรือไขกระดูก
ระบบการจัดระยะความเสี่ยงต่อเนื้องอกของระบบประสาทนานาชาติ (INRGSS)
INRGSS ใช้ปัจจัยเสี่ยงที่กำหนดโดยภาพ (IDRF) ซึ่งเป็นปัจจัยที่เห็นในการทดสอบภาพซึ่งอาจหมายความว่าเนื้องอกจะกำจัดออกได้ยากขึ้น
INRGSS แบ่ง neuroblastomas ออกเป็น 4 ระยะ:
- L1: เนื้องอกยังไม่แพร่กระจายจากจุดเริ่มต้นและยังไม่เติบโตเป็นโครงสร้างที่สำคัญ เนื้องอกจำกัดอยู่ในส่วนใดส่วนหนึ่งของร่างกาย เช่น คอ หน้าอก หรือช่องท้อง
- L2: เนื้องอกไม่ได้แพร่กระจาย (แพร่กระจาย) ไปไกลจากจุดเริ่มต้น (ตัวอย่างเช่น อาจเติบโตจากด้านซ้ายของช่องท้องไปยังด้านซ้ายของหน้าอก) แต่มี IDRF อย่างน้อยหนึ่งรายการ
- M: เนื้องอกได้แพร่กระจายไปยังส่วนที่อยู่ห่างไกลของร่างกาย (ยกเว้นเนื้องอกในระยะ MS)
- MS: โรคแพร่กระจายในเด็กอายุต่ำกว่า 18 เดือน โดยมะเร็งได้แพร่กระจายไปที่ผิวหนัง ตับ และ/หรือไขกระดูกเท่านั้น
ภาวะแทรกซ้อนและผลกระทบ
โรคมะเร็งต่อมหมวกไตมีลักษณะเฉพาะคือภาวะแทรกซ้อนและผลที่ตามมา เช่น:
- แพร่กระจาย (metastasis) ไปยังต่อมน้ำเหลือง ไขกระดูก ตับ ผิวหนัง และกระดูก
- การกดทับไขสันหลัง (ซึ่งอาจทำให้เกิดความเจ็บปวดและนำไปสู่การเป็นอัมพาต)
- การพัฒนาของโรคพารานีโอพลาสติก (เนื่องมาจากการกระทำของสารเคมีบางชนิดที่หลั่งออกมาจากเนื้องอก เช่นเดียวกับแอนติเจน disialoganglioside GD2 ที่แสดงออกโดยเซลล์ของเนื้องอก) ซึ่งแสดงออกมาโดยการเคลื่อนไหวของลูกตาอย่างรวดเร็วโดยไม่ได้ตั้งใจ การประสานงานบกพร่อง ตะคริวกล้ามเนื้อ และท้องเสีย
- อาการกำเริบหลังจากการบำบัดหลักเสร็จสิ้น (จากการปฏิบัติทางคลินิกพบว่าเนื้องอกของระบบประสาทที่มีความเสี่ยงสูงมักมีอาการกำเริบใน 50% ของกรณี)
การวินิจฉัย เนื้องอกของระบบประสาท
การวินิจฉัยโรคมะเร็งต่อมหมวกไตที่สงสัยในเด็กต้องได้รับการตรวจ ทดสอบทางห้องปฏิบัติการ และการตรวจด้วยภาพ
การตรวจเลือดและปัสสาวะจะทำเพื่อตรวจหาคาเทโคลามีน (นอร์เอพิเนฟรินและโดพามีน) และกรดโฮโมวานิลลิกหรือกรดวานิลลิลแมนเดลิก (เกิดขึ้นระหว่างการเผาผลาญของฮอร์โมนเหล่านี้) การตรวจเลือดเพื่อหาเอนไซม์อีโนเลสเฉพาะระบบประสาท การตรวจเอนไซม์เชื่อมโยงการดูดซับภูมิคุ้มกัน (ELISA) ของซีรั่มในเลือด และการวิเคราะห์ไขกระดูก (โดยเก็บตัวอย่างด้วยการเจาะดูด) จะทำการทดสอบดีเอ็นเอเพื่อระบุการกลายพันธุ์ และทำการตรวจชิ้นเนื้อเพื่อตรวจสอบไซโทมอร์โฟโลยีของเนื้อเยื่อเนื้องอก
หลังจากเก็บตัวอย่างชิ้นเนื้อแล้ว ตัวอย่างจะถูกส่งไปที่ห้องแล็ปเพื่อให้พยาธิแพทย์ (แพทย์ที่ได้รับการฝึกอบรมพิเศษในการระบุเซลล์มะเร็ง) ตรวจดูภายใต้กล้องจุลทรรศน์ นอกจากนี้ มักมีการตรวจพิเศษในห้องปฏิบัติการกับตัวอย่างเพื่อตรวจสอบว่าเนื้องอกเป็นเนื้องอกของเซลล์ประสาทหรือไม่
หากเป็นเนื้องอกของระบบประสาท การตรวจทางห้องปฏิบัติการสามารถช่วยระบุได้ว่าเนื้องอกจะเติบโตหรือแพร่กระจายได้เร็วแค่ไหน รวมถึงวิธีการรักษาแบบใดที่อาจได้ผลดีที่สุด
การวินิจฉัยด้วยเครื่องมือจะทำให้เห็นเนื้องอกได้โดยใช้คลื่นอัลตราซาวนด์ เอกซเรย์ MRI หรือ CT, PET พร้อมกับการแนะนำการสแกน 18F-fluorodeoxyglucose หรือ MIBG - การตรวจด้วยภาพด้วยเมไทโอโดเบนซิลกัวนิดีน [ 1 ]
การวินิจฉัยที่แตกต่างกัน
การวินิจฉัยแยกโรค ได้แก่ ganglioneuroma ชนิดไม่ร้ายแรง, ganglioneuroblastoma, rhabdomyosarcoma, nephroblastoma
ใครจะติดต่อได้บ้าง?
การรักษา เนื้องอกของระบบประสาท
การรักษาโรคมะเร็งต่อมหมวกไตนั้นขึ้นอยู่กับกลุ่มเสี่ยงของผู้ป่วย (ระยะของกระบวนการเกิดเนื้องอก) ตำแหน่งของเนื้องอก ลักษณะทางพันธุกรรมของเซลล์เนื้องอก และอายุของเด็ก และอาจรวมถึงการติดตามผล การผ่าตัด การให้เคมีบำบัด การฉายรังสี ภูมิคุ้มกันบำบัด และการปลูกถ่ายเซลล์ต้นกำเนิดเม็ดเลือด
การให้เคมีบำบัดแบบนีโอแอดจูแวนต์หรือแบบเสริม (ก่อนหรือหลังการผ่าตัด) สำหรับเนื้องอกของระบบประสาทในเด็ก เช่นเดียวกับการให้เคมีบำบัดสำหรับมะเร็ง อื่นๆ จะให้เป็นระยะๆ โดยให้ยาติดต่อกันหลายวัน แล้วพักการให้ยาเพื่อให้ร่างกายฟื้นตัว โดยปกติจะให้ซ้ำทุก 3-4 สัปดาห์
ยาต่อไปนี้ (และการใช้ยาผสมกัน): ไซโคลฟอสฟาไมด์, ซิสแพลตินหรือคาร์โบแพลติน, ด็อกโซรูบิซิน (เอเดรียไมซิน), วินคริสทีน, เอโทโพไซด์
ผลข้างเคียงที่พบบ่อยของยาเคมีบำบัดได้แก่ ผมร่วง เบื่ออาหาร อ่อนเพลีย คลื่นไส้และอาเจียน แผลในปาก ท้องเสีย หรือท้องผูก เคมีบำบัดสามารถทำลายไขกระดูกและทำให้จำนวนเม็ดเลือดลดลง
ภูมิคุ้มกันบำบัดแบบกำหนดเป้าหมาย (มุ่งเป้าไปที่แอนติเจนของเนื้องอก GD2) ใช้ยาจากกลุ่มแอนติบอดีโมโนโคลนอล (แอนติ-GD2 MAb) ไดนูทูซิแมบ (ยูนิทูซิน) และแนกซิทาแมบ โดยให้ยาทางเส้นเลือดดำโดยการให้ยาเป็นเวลานาน ร่วมกับตัวกระตุ้นการสร้างเม็ดเลือดขาวแบบเม็ดเลือดขาว-แมโครฟาจ (ไซโตไคน์ GM-CSF) และอินเตอร์ลิวคิน-2
ผลข้างเคียงของยาเหล่านี้ ได้แก่ อาการปวด (มักจะรุนแรงมาก) ความดันโลหิตลดลง อัตราการเต้นของหัวใจเพิ่มขึ้น หายใจถี่ (อาจมีอาการบวมของทางเดินหายใจ) อุณหภูมิร่างกายสูงขึ้น คลื่นไส้ อาเจียน และท้องเสีย การเปลี่ยนแปลงในองค์ประกอบของเซลล์และแร่ธาตุในเลือด
เพื่อลดความเสี่ยงของการเกิดมะเร็งซ้ำหลังการให้เคมีบำบัดขนาดสูงและการปลูกถ่ายเซลล์ต้นกำเนิด เด็กที่เป็นโรคมะเร็งต่อมหมวกไตที่มีความเสี่ยงสูงจะได้รับการรักษาด้วยเรตินอยด์แบบระบบ กรด 13-ซิส-เรตินอยด์ (ไอโซเตรติโนอิน) [ 2 ]
การรักษาทางศัลยกรรมสำหรับเนื้องอกของต่อมหมวกไต เช่น การผ่าตัดต่อมหมวกไตแบบเปิดหรือการผ่าตัดผ่านกล้องสำหรับเนื้องอกของต่อมหมวกไตการผ่าตัดต่อมน้ำเหลือง (การเอาต่อมน้ำเหลืองที่ได้รับผลกระทบออก) เป็นต้น [ 3 ]
สำหรับโรคมะเร็งต่อมหมวกไตที่มีความเสี่ยงสูงอาจใช้การรักษาด้วยรังสี[ 4 ]
การป้องกัน
เมื่อพิจารณาถึงสาเหตุของเนื้องอกของเซลล์ประสาทในเด็ก การป้องกันที่ดีที่สุดคือการปรึกษาทางพันธุกรรมเมื่อวางแผนตั้งครรภ์ แต่ควรทราบไว้ว่าเนื้องอกนี้มีความเกี่ยวข้องกับการกลายพันธุ์ที่ถ่ายทอดทางพันธุกรรมเพียง 1-2% ของกรณีเท่านั้น
พยากรณ์
เนื้องอกของระบบประสาทในวัยทารกมีศักยภาพที่จะกลับเป็นปกติได้เอง
เครื่องหมายการพยากรณ์โรค
- เนื้องอกที่มีความเสี่ยงสูง เช่นเดียวกับเนื้องอกของต่อมหมวกไตในเด็กทุกกลุ่มอายุและทุกระยะ (ยกเว้นระยะ 4S) ซึ่งมีการแสดงออกของยีน N-MYC ที่เพิ่มขึ้นและการขยายตัวของยีนก่อมะเร็ง N-Myc มีการพยากรณ์โรคที่ไม่ดีซึ่งส่งผลต่ออายุขัย
- การมีเซลล์เนื้องอกที่ขาดหายไปบางส่วนของโครโมโซม 1 หรือ 11 (เรียกว่า 1p หรือ 11q deletions) จะทำให้การพยากรณ์โรคแย่ลง การมีโครโมโซม 17 ส่วนเกิน (17q gain) ยังทำให้การพยากรณ์โรคแย่ลงด้วย
- เซลล์เนื้องอกของระบบประสาทที่มี DNA จำนวนมากจะมีการพยากรณ์โรคที่ดีกว่า โดยเฉพาะในเด็กอายุต่ำกว่า 2 ปี
- เนื้องอกของระบบประสาทที่มีตัวรับนิวโรโทรฟินมากขึ้น โดยเฉพาะอย่างยิ่งตัวรับปัจจัยการเจริญเติบโตของเส้นประสาท TrkA จะมีการพยากรณ์โรคที่ดีกว่า
การอยู่รอดตามกลุ่มเสี่ยง Childhood Oncology Group (COG)
- กลุ่มเสี่ยงต่ำ: เด็กในกลุ่มเสี่ยงต่ำมีอัตราการมีชีวิตรอด 5 ปีมากกว่า 95%
- กลุ่มเสี่ยงปานกลาง: เด็กในกลุ่มเสี่ยงปานกลางมีอัตราการรอดชีวิต 5 ปีที่ 90% ถึง 95%
- กลุ่มเสี่ยงสูง: เด็กในกลุ่มเสี่ยงสูงมีอัตราการมีชีวิตรอด 5 ปีประมาณ 50%
ประมาณ 15% ของการเสียชีวิตจากมะเร็งในวัยเด็กเกิดจากเนื้องอกของระบบประสาท โอกาสที่มะเร็งที่มีความเสี่ยงสูงนี้จะมีชีวิตรอดในระยะยาวไม่เกิน 40% อัตราการรอดชีวิตโดยรวมใน 5 ปีอยู่ที่ 67-74% โดย 43% อยู่ในกลุ่มอายุ 1-4 ปี และมากกว่า 80% สำหรับเนื้องอกของระบบประสาทที่ได้รับการวินิจฉัยในปีแรกของชีวิต
Использованная литература