ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
โรค Charcot-Marie-Tooth
ตรวจสอบล่าสุด: 23.11.2021

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
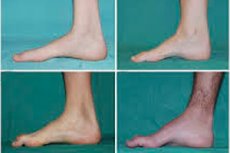
โรคกล้ามเนื้อช่องท้องฝ่อดาวน์ซินโดรมหรือโรค Charcot-Marie-Tooth เป็นโรคทางพันธุกรรมเรื้อรังทั้งหมดที่มีความเสียหายต่อเส้นประสาทส่วนปลาย
ตาม ICD-10 ในส่วนของโรคของระบบประสาทรหัสของโรคนี้คือ G60.0 (มอเตอร์ทางพันธุกรรมและโรคระบบประสาทประสาทสัมผัส) นอกจากนี้ยังรวมอยู่ในรายชื่อโรคเด็กกำพร้า
ระบาดวิทยา
ตามสถิติทางคลินิกความชุกของโรค Charcot-Marie-Tooth ทุกประเภทต่อประชากร 100,000 คนอยู่ที่ 19 ราย (ตามแหล่งข้อมูลอื่นหนึ่งกรณีต่อประชากร 2.5-10,000 คน)
CMT type 1 คิดเป็นประมาณสองในสามของผู้ป่วย (หนึ่งกรณีต่อประชากร 5-7 พันคน) และเกือบ 70% มีความเกี่ยวข้องกับการทำซ้ำยีน PMP22 ในโลกโรคประเภทนี้ส่งผลกระทบต่อผู้คนมากกว่า 1.2 ล้านคน
อุบัติการณ์ของ CMT ประเภท 4 ประมาณ 1-5 รายต่อเด็ก 10,000 คน [1]
สาเหตุ โรค Charcot-Marie-Tooth
ตามการ จำแนกประเภทของกลุ่มอาการ polyneuropathic การฝ่อของกล้ามเนื้อในช่องท้อง(peroneal) การเสื่อมสภาพของระบบประสาท Charcot-Marie-Tooth หรือโรค Charcot-Marie-Tooth (ย่อว่า CMT) หมายถึง polyneuropathies ทางประสาทสัมผัสทางมอเตอร์ที่กำหนดโดยทางพันธุกรรม [2]
นั่นคือสาเหตุของการเกิดขึ้นคือการกลายพันธุ์ทางพันธุกรรม และขึ้นอยู่กับลักษณะของความผิดปกติทางพันธุกรรมประเภทหลักหรือประเภทของกลุ่มอาการนี้แตกต่างกันไป: demyelinating และ axonal กลุ่มแรกรวมถึงโรค Charcot-Marie-Tooth ชนิดที่ 1 (CMT1) ซึ่งเกิดขึ้นจากการทำสำเนายีน PMP22 บนโครโมโซม 17 ซึ่งเข้ารหัสโปรตีนไมอีลินส่วนปลายของ transmembrane 22 เป็นผลให้การแยกส่วนของเปลือกแอกซอน (กระบวนการของเซลล์ประสาท) และการลดลงของความเร็วในการนำกระแสประสาทเกิดขึ้นสัญญาณ นอกจากนี้อาจมีการกลายพันธุ์ในยีนอื่นบางยีน
รูปแบบแอกซอนคือ Charcot-Marie-Tooth disease type 2 (CMT2) ซึ่งมีผลต่อแอกซอนและเกี่ยวข้องกับการเปลี่ยนแปลงทางพยาธิวิทยาในยีน MFN2 ที่ตำแหน่ง 1p36.22 ซึ่งเข้ารหัสโปรตีนเมมเบรน mitofusin-2 ซึ่งจำเป็น สำหรับ mitochondrial fusion และการก่อตัวของเครือข่าย mitochondrial ที่ทำงานภายในเซลล์ประสาทส่วนปลาย CMT2 มีชนิดย่อยมากกว่าหนึ่งโหล (ที่มีการกลายพันธุ์ในยีนเฉพาะ)
ควรสังเกตว่าขณะนี้มีการระบุยีนมากกว่าร้อยยีนซึ่งความเสียหายที่ถ่ายทอดทางพันธุกรรมทำให้เกิดโรค Charcot-Marie-Tooth ชนิดย่อยต่างๆ ตัวอย่างเช่นการกลายพันธุ์ในยีน RAB7 จะพัฒนาชนิด 2B CMT; การเปลี่ยนแปลงของยีน SH3TC2 (ซึ่งเข้ารหัสโปรตีนชนิดใดชนิดหนึ่งของเยื่อหุ้มเซลล์ Schwann) ทำให้เกิดชนิด 4C CMT ซึ่งปรากฏตัวในวัยเด็กและมีลักษณะการสลายตัวของมอเตอร์และเซลล์ประสาทรับความรู้สึก (รูปแบบที่ 4 หนึ่งและครึ่งโหลของ โรคนี้มีความโดดเด่น)
SMT ประเภท 3 ที่หายาก (เรียกว่า Dejerine-Sott syndrome) ซึ่งเกิดจากการกลายพันธุ์ในยีน PMP22, MPZ, EGR2 และอื่น ๆ ก็เริ่มพัฒนาในเด็กปฐมวัย
เมื่อ CMT type 5 เกิดขึ้นเมื่ออายุ 5-12 ปีไม่เพียง แต่เป็นโรคระบบประสาท (ในรูปแบบของอาการกระตุกของแขนขาด้านล่าง) เท่านั้น แต่ยังสร้างความเสียหายต่อประสาทตาและประสาทหูด้วย
กล้ามเนื้ออ่อนแรงและเส้นประสาทตาฝ่อ (สูญเสียการมองเห็น) รวมทั้งปัญหาเกี่ยวกับการทรงตัวเป็นลักษณะของ CMT type 6 และด้วยโรค Charcot-Marie-Tooth ประเภทที่ 7 ไม่เพียง แต่สังเกตเห็นโรคระบบประสาทประสาทสัมผัสเท่านั้น แต่ยังรวมถึงโรคจอประสาทตาในรูปแบบของ retinitis pigmentosa ด้วย
โรค SMT ที่เชื่อมโยงกับ X หรือ Charcot-Marie-Tooth ที่พบได้บ่อยกว่าที่มี tetraparesis ของแขนขา (การเคลื่อนไหวของแขนและขาทั้งสองข้างที่อ่อนแอลง) ในผู้ชายเป็นชนิดที่แยกไม่ออกและถือว่าเป็นผลมาจากการกลายพันธุ์ของยีน GJB1 ใน แขนยาวของโครโมโซม X ซึ่งเป็นรหัสสำหรับคอนเน็กซิน 32 ซึ่งเป็นโปรตีนจากเซลล์ Schwann และเซลล์โอลิโกเดนโดรไซท์ซึ่งควบคุมการส่งสัญญาณประสาท [3]
ปัจจัยเสี่ยง
ปัจจัยเสี่ยงหลักของ CMT คือการปรากฏตัวของโรคนี้ในประวัติครอบครัวนั่นคือในญาติสนิท
ตามที่นักพันธุศาสตร์ระบุว่าหากทั้งพ่อและแม่เป็นพาหะของยีนด้อยถอยอัตโนมัติของโรค Charcot-Marie-Tooth ความเสี่ยงของการมีบุตรที่จะเป็นโรคนี้คือ 25% และความเสี่ยงที่เด็กจะมียีนนี้ (แต่ตัวเขาเองจะไม่มีอาการใด ๆ ) อยู่ที่ประมาณ 50%
ในกรณีของการถ่ายทอดทางพันธุกรรม X-linked (เมื่อยีนที่กลายพันธุ์อยู่บนโครโมโซม X ของผู้หญิง) มีความเสี่ยง 50% ที่แม่จะส่งต่อยีนนี้ไปยังลูกชายของเธอและเขาจะเป็นโรค CMT เมื่อเด็กหญิงเกิดมาโรคนี้อาจไม่เกิดขึ้น แต่ลูกชายของลูกสาว (หลาน) สามารถสืบทอดยีนที่บกพร่องได้ - ด้วยพัฒนาการของโรค
กลไกการเกิดโรค
ในโรค Charcot-Marie-Tooth ทุกประเภทการเกิดโรคเกิดจากความผิดปกติทางพันธุกรรมของเส้นประสาทส่วนปลาย: มอเตอร์ (มอเตอร์) และประสาทสัมผัส (ประสาทสัมผัส)
หากชนิด CMT มีการสลายตัวการทำลายหรือความบกพร่องของปลอกไมอีลินซึ่งปกป้องแอกซอนของเส้นประสาทส่วนปลายจะนำไปสู่การชะลอตัวในการส่งกระแสประสาทของระบบประสาทส่วนปลาย - ระหว่างสมองกล้ามเนื้อและอวัยวะรับความรู้สึก.
ในโรคประเภทแอกซอนแอกซอนได้รับผลกระทบโดยตรงซึ่งส่งผลเสียต่อความแรงของสัญญาณประสาทซึ่งไม่เพียงพอสำหรับการกระตุ้นกล้ามเนื้อและอวัยวะรับสัมผัสอย่างเต็มที่
อ่านเพิ่มเติม:
Charcot-Marie-Tooth syndrome แพร่กระจายอย่างไร? ยีนที่มีข้อบกพร่องสามารถถ่ายทอดทางพันธุกรรมได้ในลักษณะถอยกลับอัตโนมัติหรือ autosomal recessive
สิ่งที่พบบ่อยที่สุด - การถ่ายทอดทางพันธุกรรมที่โดดเด่นของ autosomal - เกิดขึ้นเมื่อมียีนที่กลายพันธุ์หนึ่งสำเนา (ดำเนินการโดยพ่อแม่คนใดคนหนึ่ง) และความน่าจะเป็นของการแพร่เชื้อ CMT ไปยังเด็กแต่ละคนที่เกิดนั้นอยู่ที่ประมาณ 50% [4]
ในการถ่ายทอดทางพันธุกรรมแบบถอยโดยอัตโนมัติโรคนี้ต้องการสำเนาของยีนที่บกพร่องสองชุด (หนึ่งชุดจากพ่อแม่แต่ละคนที่ไม่มีอาการของโรค)
ใน 40-50% ของกรณีการลอกเลียนแบบทางพันธุกรรมที่โดดเด่นของ autosomal เกิดขึ้นนั่นคือ CMT type 1; ใน 12-26% ของกรณี - Axonal CMT นั่นคือประเภท 2 และใน 10-15% ของกรณีจะสังเกตเห็นการถ่ายทอดทางพันธุกรรมที่เชื่อมโยงกับ X [5]
อาการ โรค Charcot-Marie-Tooth
โดยปกติสัญญาณแรกของโรคนี้จะเริ่มปรากฏในวัยเด็กและวัยรุ่นและค่อยๆพัฒนาไปตลอดชีวิตแม้ว่ากลุ่มอาการนี้จะทำให้รู้สึกได้ในภายหลัง การรวมกันของอาการมีความแปรปรวนและไม่สามารถคาดการณ์อัตราการลุกลามของโรครวมทั้งความรุนแรงได้
มีอาการทั่วไปในระยะเริ่มต้นเช่นความเหนื่อยล้าทั่วไปที่เพิ่มขึ้น ลดเสียง (ความอ่อนแอ) ของกล้ามเนื้อเท้าข้อเท้าและขาส่วนล่าง ขาดการตอบสนอง ทำให้ขยับเท้าได้ยากและนำไปสู่ dysbasia (การเดินผิดปกติ) ในรูปแบบของการยกขาที่สูงขึ้นซึ่งมักมีการสะดุดและล้มบ่อย อาการของโรคคอ-Marie-ฟันในเด็กเล็กอาจจะเด่นชัดซุ่มซ่ามและความยากลำบากเดินผิดปกติสำหรับอายุที่เกี่ยวข้องกับ การเดินเท้าห้อยทวิภาคี ความผิดปกติของเท้าก็มีลักษณะเช่นกัน: ส่วนโค้งสูง (เท้ากลวง) หรือเท้าแบนแข็งแรงนิ้วโค้ง (เหมือนค้อน)
ในกรณีที่เดินด้วยปลายเท้ากับพื้นหลังของความดันเลือดต่ำของกล้ามเนื้อนักประสาทวิทยาอาจสงสัยว่าเด็กมี CMT type 4 ซึ่งเด็กในวัยรุ่นอาจไม่สามารถเดินได้
ในขณะที่ดำเนินไปกล้ามเนื้อลีบและอ่อนแรงจะลามไปที่ปลายแขนทำให้ทักษะการเคลื่อนไหวดีและกิจกรรมมือตามปกติทำได้ยาก การลดลงของความรู้สึกสัมผัสและความสามารถในการรู้สึกอบอุ่นและเย็นรวมถึงอาการชาที่เท้าและมือบ่งบอกถึงความเสียหายต่อแอกซอนของเส้นประสาทรับความรู้สึก
เมื่อปรากฏในวัยเด็กของโรค Charcot-Marie-Tooth ประเภทที่ 3 และ 6 จะมีอาการ ataxia ที่ไว (การประสานการเคลื่อนไหวและการทรงตัวบกพร่อง) การกระตุกของกล้ามเนื้อและการสั่นสะเทือนความเสียหายต่อเส้นประสาทบนใบหน้าฝ่อประสาทตาที่มีอาตาการสูญเสียการได้ยิน
ในระยะต่อมาอาจมีอาการสั่นที่ไม่สามารถควบคุมได้ (ตัวสั่น) และปวดกล้ามเนื้อบ่อยๆ ปัญหาเกี่ยวกับการเคลื่อนไหวอาจนำไปสู่การพัฒนาความเจ็บปวด: กล้ามเนื้อข้อต่อระบบประสาท
ภาวะแทรกซ้อนและผลกระทบ
โรค Charcot-Marie-Tooth อาจมีภาวะแทรกซ้อนและผลกระทบเช่น:
- เคล็ดขัดยอกและกระดูกหักบ่อยขึ้น
- การหดตัวที่เกี่ยวข้องกับการสั้นลงของกล้ามเนื้อและเส้นเอ็นรอบนอก
- scoliosis (ความโค้งของกระดูกสันหลัง);
- ปัญหาการหายใจ - มีความเสียหายต่อเส้นใยประสาทที่ทำให้กล้ามเนื้อของไดอะแฟรมอยู่ภายใน:
- การสูญเสียความสามารถในการเคลื่อนที่อย่างอิสระ
การวินิจฉัย โรค Charcot-Marie-Tooth
การวินิจฉัยรวมถึงการตรวจทางคลินิกประวัติ (รวมถึงประวัติครอบครัว) การตรวจระบบประสาทและระบบ
การทดสอบจะดำเนินการเพื่อตรวจสอบช่วงของการเคลื่อนไหวความไวและการตอบสนองของเอ็น การนำกระแสประสาทสามารถประเมินได้โดยการวินิจฉัยเครื่องมือ - ไฟฟ้าหรือ electroneuromyography อาจต้องใช้อัลตราซาวนด์หรือ MRI [6]
การวินิจฉัยทางพันธุกรรมหรือดีเอ็นเอเพื่อระบุการกลายพันธุ์ทางพันธุกรรมที่พบบ่อยที่สุดที่ทำให้เกิด CMT ในตัวอย่างเลือดมีข้อ จำกัด เนื่องจากการตรวจดีเอ็นเอยังไม่สามารถใช้ได้กับ CMT ทุกประเภท ดูรายละเอียด - การวิจัยทางพันธุกรรม
ในบางกรณีการตรวจชิ้นเนื้อของเส้นประสาทส่วนปลาย (โดยปกติคือ gastrocnemius)
การวินิจฉัยที่แตกต่างกัน
การวินิจฉัยแยกโรคจะดำเนินการกับโรคระบบประสาทส่วนปลายอื่น ๆ โรคกล้ามเนื้อ Duchenne กล้ามเนื้อเสื่อมและ myelopathic และ myasthenic โรคระบบประสาทอักเสบจากโรคเบาหวานร่วมกับ myeloaptias ในกรณีของเส้นโลหิตตีบด้านข้างหลายเส้นและ amyotrophic, Guillain-Barré syndrome, การบาดเจ็บของเส้นประสาทส่วนปลายและกระดูกสันหลังของดิสก์ ) ความเสียหายต่อสมองน้อยหรือฐานดอกตลอดจนผลข้างเคียงของเคมีบำบัด (เมื่อรับการรักษาด้วย cytostatics เช่น Vincristine หรือ Paclitaxel) [7]
ใครจะติดต่อได้บ้าง?
การรักษา โรค Charcot-Marie-Tooth
วันนี้การรักษาโรคทางพันธุกรรมนี้ประกอบด้วยแบบฝึกหัดกายภาพบำบัด (มุ่งเป้าไปที่การเสริมสร้างและยืดกล้ามเนื้อ) กิจกรรมบำบัด (ซึ่งช่วยให้ผู้ป่วยกล้ามเนื้ออ่อนแรงในมือ); ใช้อุปกรณ์เกี่ยวกับกระดูกเพื่อความสะดวกในการเดิน หากจำเป็นให้รับประทานยาแก้ปวดหรือยากันชัก [8]
ในกรณีของเท้าแบนที่เด่นชัดสามารถทำการผ่าตัดกระดูกได้และในกรณีที่ส้นเท้าเสียรูปจะมีการระบุการแก้ไขด้วยการผ่าตัด - arthrodesis [9]
กำลังดำเนินการวิจัยเกี่ยวกับองค์ประกอบทางพันธุกรรมของโรคและวิธีการรักษา การใช้เซลล์ต้นกำเนิดฮอร์โมนบางชนิดเลซิตินหรือกรดแอสคอร์บิกยังไม่ให้ผลลัพธ์ที่เป็นบวก
แต่ต้องขอบคุณการวิจัยล่าสุดในอนาคตอันใกล้นี้อาจมีการวิจัยใหม่ในการรักษาโรค Charcot-Marie-Tooth ดังนั้นตั้งแต่ปี 2014 บริษัท Pharnext ของฝรั่งเศสได้ทำการพัฒนาและตั้งแต่กลางปี 2019 การทดลองทางคลินิกของยา PXT3003 สำหรับการรักษา CMT type 1 ในผู้ใหญ่ยับยั้งการแสดงออกที่เพิ่มขึ้นของยีน PMP22 ปรับปรุงการไมอีไลของเส้นประสาทส่วนปลายและ อาการทางประสาทและกล้ามเนื้ออ่อนแอลง
ผู้เชี่ยวชาญของ บริษัท การแพทย์ Sarepta Therapeutics (USA) กำลังดำเนินการเกี่ยวกับยีนบำบัดสำหรับโรค Charcot-Marie-Tooth ประเภท 1 การบำบัดนี้จะใช้ไวรัสที่เกี่ยวข้องกับอะดีโน (AAV) ที่ไม่เป็นอันตรายของสกุล Dependovirus ที่มีจีโนมดีเอ็นเอแบบเส้นเดียวเชิงเส้นซึ่งจะนำยีน NTF3 เข้าสู่ร่างกายซึ่งจะเข้ารหัสโปรตีน neurotrophin-3 (NT-3) ที่จำเป็นสำหรับ การทำงานของเซลล์ประสาท Schwann
ภายในสิ้นปี 2020 Helixmith จะเริ่มการทดลองทางคลินิกของการบำบัดด้วยยีน Engensis (VM202) ที่พัฒนาขึ้นในเกาหลีใต้เพื่อรักษาอาการของกล้ามเนื้อใน CMT ประเภท 1 [10]
การป้องกัน
การป้องกัน CMT อาจเป็นการให้คำปรึกษาทางพันธุกรรมของพ่อแม่ในอนาคตโดยเฉพาะอย่างยิ่งหากมีคนจากคู่แต่งงานเป็นโรคนี้ในครอบครัว อย่างไรก็ตามมีการระบุกรณีของการกลายพันธุ์ของยีน de novo นั่นคือในกรณีที่ไม่มีโรคในประวัติครอบครัว
ในระหว่างตั้งครรภ์การสุ่มตัวอย่าง chorionic villus (อายุครรภ์ 10 ถึง 13 สัปดาห์) รวมทั้งการวิเคราะห์น้ำคร่ำ (ที่ 15-18 สัปดาห์) ช่วยให้คุณสามารถตรวจสอบโอกาสในการเกิดโรค Charcot-Marie-Tooth ในเด็กในครรภ์ได้
พยากรณ์
โดยทั่วไปการพยากรณ์โรคสำหรับโรค Charcot-Marie-Tooth ประเภทต่างๆขึ้นอยู่กับความรุนแรงทางคลินิก แต่ไม่ว่าในกรณีใดโรคจะดำเนินไปอย่างช้าๆ ผู้ป่วยจำนวนมากมีความพิการแม้ว่าจะไม่ได้ลดอายุขัย