ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
โรคไตอักเสบทางพันธุกรรม (Alport syndrome) ในเด็ก
ตรวจสอบล่าสุด: 23.04.2024

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
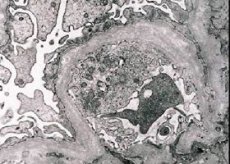
โรคไตอักเสบพันธุกรรม (กลุ่มอาการอัลพอร์ต) - กำหนดทางพันธุกรรมที่ไม่ได้รับการถ่ายทอดภูมิคุ้มกัน glomerulopathy exhibiting ปัสสาวะเป็นเลือด (บางครั้งโปรตีน) ลดลงความก้าวหน้าในการทำงานของไตกับการพัฒนาของไตวายเรื้อรังที่มักจะรวมกับหูหนวกประสาทและความผิดปกติของการมองเห็น.
เป็นครั้งแรกที่โรคได้อธิบายไว้ 1902 г. L.G.Guthrie, ผู้สังเกตครอบครัวในหลายชั่วอายุคนซึ่งเป็นที่สังเกตเห็นโลหิตจาง ใน 1915 у สมาชิกในครอบครัวเดียวกัน A.F.Hurst อธิบายการพัฒนาของอุรีเรีย ใน 1927 г. A Alport เป็นครั้งแรกเผยให้เห็นการสูญเสียการได้ยินในญาติหลายคนที่มี gematuriya ใน 50 ปีของศตวรรษที่ผ่านมาได้อธิบายความพ่ายแพ้ของตาในโรคที่คล้ายกัน ใน 1972 г. у ผู้ป่วยที่มีเลือดออกทางพันธุกรรมในการศึกษาเกี่ยวกับรูปร่างของเนื้อเยื่อไต Hinglais et al. เผยให้เห็นการขยายตัวที่ไม่สม่ำเสมอและการแยกส่วนของเยื่อหุ้มสมองกลวง ใน 1985 г. พื้นฐานทางพันธุกรรมของโรคไตอักเสบทางพันธุกรรมได้รับการระบุ - การกลายพันธุ์ในยีนของคอลลาเจนชนิด IV (Fiengold et al., 1985).
การตรวจสอบลักษณะทางพันธุกรรมของโรคทำให้สามารถสรุปได้ว่าความแตกต่างในการแสดงออกทางฟีโนไทป์ของโรคไตอักเสบทางพันธุกรรม (ที่มีหรือไม่มีการสูญเสียการได้ยิน) เป็นเพราะระดับของการแสดงออกของยีนที่กลายพันธุ์ ดังนั้นในปัจจุบันตัวแปรทางคลินิกทั้งหมดจะถือว่าเป็นอาการของโรคหนึ่งและคำว่า "โรคไตอักเสบทางพันธุกรรม" เป็นความหมายเหมือนกันกับคำว่า "Alport syndrome.".
ตามการศึกษาด้านระบาดวิทยาโรคไตอักเสบทางพันธุกรรมเกิดขึ้นที่ความถี่ 17 100 000 ประชากรเด็ก.
สาเหตุของโรค Alport
พื้นฐานทางพันธุกรรมของโรคคือการกลายพันธุ์ของยีน a-5 ของสายโซ่คอลลาเจนชนิดที่ 4 ประเภทของสากลสำหรับเยื่อฐานไตอุปกรณ์ประสาทหูแคปซูลเลนส์จอประสาทตาและกระจกตาที่แสดงในการศึกษาโดยใช้โคลนอลแอนติบอดีกับส่วนคอลลาเจน เมื่อเร็ว ๆ นี้พวกเขาระบุความเป็นไปได้ในการใช้เครื่องตรวจดีเอ็นเอสำหรับการวินิจฉัยก่อนคลอดของโรคไตอักเสบทางพันธุกรรม
ความสำคัญของการทดสอบสมาชิกทุกคนในครอบครัวโดยใช้ DNA probes เพื่อระบุผู้ให้บริการยีนที่กลายพันธุ์จะเน้นซึ่งเป็นสิ่งที่สำคัญมากในการให้คำปรึกษาทางพันธุกรรมทางการแพทย์ของครอบครัวที่มีโรคนี้ อย่างไรก็ตามถึง 20% ของครอบครัวไม่มีญาติที่เป็นโรคไตซึ่งแสดงให้เห็นว่ามีอุบัติการณ์การกลายพันธุ์ที่เกิดขึ้นเองในยีนที่ผิดปกติสูง ผู้ป่วยส่วนใหญ่ที่เป็นโรคไตอักเสบทางพันธุกรรมในครอบครัวมีบุคคลที่เป็นโรคไตการสูญเสียการได้ยินและวิสัยทัศน์ทางพยาธิวิทยา การแต่งงานระหว่างคนที่มีบรรพบุรุษอย่างน้อยหนึ่งคนเนื่องจากการแต่งงานของบุคคลที่เกี่ยวข้องจะเพิ่มโอกาสในการได้ยีนเดียวกันจากพ่อแม่ทั้งสองคน มีการสร้าง autosomal dominant และ autosomal recessive และ dominant ซึ่งเชื่อมโยงกับโครโมโซม X ของเส้นทางการส่งผ่าน
เด็กมีแนวโน้มที่จะแยกแยะความแตกต่างของสามชนิดของโรคไตอักเสบทางพันธุกรรม: Alport syndrome, โรคไตอักเสบทางพันธุกรรมโดยไม่สูญเสียการได้ยินและครอบครัวเนื้องอกที่ไม่เป็นอันตราย
Alport Syndrome -โรคไตอักเสบทางพันธุกรรมที่มีความบกพร่องในการได้ยิน พื้นฐานเป็นข้อบกพร่องรวมกันในโครงสร้างของคอลลาเจนของเยื่อหุ้มปอดของ glomeruli ของไตโครงสร้างของหูและตา ยีนของ Alport syndrome แบบคลาสสิกตั้งอยู่ที่ตำแหน่ง 21-22 q ของแขนยาวของโครโมโซม X ในกรณีส่วนใหญ่จะได้รับการสืบทอดมาจากชนิดเด่นที่เชื่อมโยงกับโครโมโซม X ในเรื่องนี้ในผู้ชายโรค Alport เป็นเรื่องยากมากขึ้นเพราะในผู้หญิงการทำงานของยีนที่กลายพันธุ์ได้รับการชดเชยโดยอัลลีลที่มีสุขภาพดีของโครโมโซมที่สองที่เหมือนกัน
พื้นฐานทางพันธุกรรมของพัฒนาการของโรคไตอักเสบทางพันธุกรรมคือการกลายพันธุ์ในยีนของกลุ่มอัลฟาของคอลลาเจนชนิด IV เป็นที่รู้จักกันหกโซ่ชนิด IV คอลลาเจน G: A5 และ A6 ยีนโซ่ (Sol4A5 และ Sol4A5) ตั้งอยู่บนแขนยาวของโครโมโซมในเขต 21-22q; ยีนของ a3- และ a4-chains (Co4A3 และ Co4A4) - ในโครโมโซม 2 nd; ยีนของ a1- และ a2-chains (Co4A1 และ Co4A2) - บนโครโมโซมที่ 13
ในกรณีส่วนใหญ่ (80-85%) การมีส่วนร่วมในการถ่ายทอดโรคของ X-linked มีความเกี่ยวข้องกับความเสียหายที่เกิดขึ้นกับยีน Co4A5 เนื่องจากการลบการกลายพันธุ์จุดหรือความผิดปกติของ splicing ปัจจุบันมีการกลายพันธุ์ของยีน Kol4A5 มากกว่า 200 ชนิดซึ่งมีส่วนรับผิดชอบต่อการละเมิดการสังเคราะห์คอลลาเจนชนิดที่ 5 ของสายโซ่ a5 ในประเภทของการถ่ายทอดทางพันธุกรรมนี้โรคแสดงออกในเด็กทั้งสองเพศ แต่ในเด็กผู้ชายมันเป็นเรื่องยากมากขึ้น
การกลายพันธุ์ของยีน Co4A3 และ Co4A4 ที่รับผิดชอบในการสังเคราะห์คอลลาเจนชนิดที่ 3 และ a4 เป็นกรรมพันธุ์ที่ได้รับการถ่ายทอดทางพันธุกรรม จากผลการวิจัยพบว่าชนิดเด่นของ autosomal inherit คือ 16% ของกรณีที่เป็นโรคไตเสื่อมทางพันธุกรรม autosomal recessive ในผู้ป่วย 6% มีการกลายพันธุ์ Co4A3 และ Co4A4 ประมาณ 10 ครั้ง
ผลของการกลายพันธุ์คือการฝ่าฝืนกระบวนการของการประกอบคอลลาเจนชนิด IV ทำให้เกิดการหยุดชะงักในโครงสร้างของมัน คอลลาเจนชนิดที่ 4 เป็นหนึ่งในองค์ประกอบหลักของเมมเบรนชั้นเยื่อหุ้มสมอง (glomerular basement membrane) อุปกรณ์ cochlear และเลนส์ตาซึ่งพยาธิวิทยาจะถูกเปิดเผยในคลินิกโรคไตอักเสบทางพันธุกรรม
คอลลาเจนชนิด IV ซึ่งเป็นส่วนหนึ่งของเยื่อชั้นใต้ดินไตประกอบด้วยหลักของสองโซ่ a1 (IV) และโซ่ a2 หนึ่ง (IV) และยังมี A3, A4, A5 ห่วงโซ่ ส่วนใหญ่มักจะเมื่อ X-linked มรดก Sol4A5 กลายพันธุ์มาพร้อมกับการขาด a3, a4- และโซ่ A5 A6 ประเภทคอลลาเจน IV ในโครงสร้างและจำนวนของ O1 และโซ่ a2 ลงไปในชั้นใต้ดินไตเยื่อหุ้มเซลล์เพิ่มขึ้น กลไกของปรากฏการณ์นี้ไม่ชัดเจนสันนิษฐานว่าสาเหตุคือการเปลี่ยนแปลงหลังการแปลใน mRNA
A3 ขาด a4- และโซ่ a5 ในเมมเบรน IV คอลลาเจนชั้นใต้ดินชนิดโครงสร้างของผล glomeruli ในการทำให้ผอมบางและความเปราะบางของขั้นเริ่มต้นของกลุ่มอาการอัลพอร์ตที่ปรากฏตัวทางคลินิกปัสสาวะเป็นเลือดมากที่สุด (บางครั้งปัสสาวะหรือโปรตีนเพียงโปรตีน) และการสูญเสีย lenticonus ได้ยิน ความก้าวหน้าต่อไปของโรคจะนำไปสู่ความหนาและการหยุดชะงักของการซึมผ่านเยื่อหุ้มเซลล์แรกเริ่มในช่วงปลายของการเกิดโรคที่มีการเจริญเติบโตในประเภทคอลลาเจนเหล่านี้วีวีและที่ประจักษ์ในการเพิ่มขึ้นของโปรตีนและลดการทำงานของไต
ลักษณะของการกลายพันธุ์ของโรคไตอักเสบในตระกูลพันธุกรรมส่วนใหญ่จะเป็นตัวกำหนดลักษณะของฟีโนไทป์ เมื่อ X ลบโครโมโซมที่มีการกลายพันธุ์ของยีนพร้อมกันและ Sol4A6 Sol4A5 รับผิดชอบสำหรับการสังเคราะห์ A5 และ A6 โซ่ของคอลลาเจนชนิด IV รวมกับซินโดรม Alport หลอดอาหาร leiomyomatosis และอวัยวะเพศ ตามการศึกษาที่มีการกลายพันธุ์ของยีนที่เกี่ยวข้องกับการ Sol4A5 ลบมีการทำเครื่องหมายความรุนแรงขนาดใหญ่ของกระบวนการทางพยาธิวิทยาร่วมกับแผลอาการไต extrarenal และการพัฒนาในช่วงต้นของไตวายเรื้อรังที่เมื่อเทียบการกลายพันธุ์ของยีน stochechnoy นี้
จลนศาสตร์ของกล้องจุลทรรศน์อิเล็กตรอนแสดงให้เห็นถึงการผอมบางและการบั่นปากของเยื่อหุ้มปอดเยื่อหุ้มปอด (โดยเฉพาะ lamina densa) และการปรากฏตัวของเม็ดมีความหนาแน่นทางอิเล็กทรอนิกส์ บาดแผลของ glomerulus อาจไม่เหมือนกันในผู้ป่วยรายเดียวกันเนื่องจากมีแผลเป็นจาก mesangium ถึง glomerulosclerosis เพียงเล็กน้อย Glomerulitis ใน Alport syndrome มักเป็นภูมิคุ้มกันซึ่งจะแยกแยะออกจาก glomerulonephritis ลักษณะคือการพัฒนาของการคลาดเคลื่อนของคลองแทรกซึม lymphohistiocyte การปรากฏตัวของ "เซลล์โฟม" กับการรวมของไขมัน - lipofagi กับความคืบหน้าของโรค, การทำลายความหนาและการทำเครื่องหมายของเยื่อหุ้มปอด glomeruli ฐานจะถูกเปิดเผย
มีการเปิดเผยการเปลี่ยนแปลงสภาพระบบภูมิคุ้มกันบางอย่าง ในผู้ป่วยที่มีโรคไตอักเสบทางพันธุกรรมระดับ IgA ลดลงและแนวโน้มที่จะเพิ่มความเข้มข้นของ IgM ในเลือดจะสังเกตได้ว่าระดับ IgG จะเพิ่มขึ้นในระยะเริ่มแรกของการเกิดโรคและลดลงในช่วงปลายเดือน บางทีการเพิ่มขึ้นของความเข้มข้นของ IgM และ G คือการตอบสนองที่ชดเชยในการตอบสนองต่อการขาดดุลของ IgA
กิจกรรมการทำงานของระบบ T-lymphocyte จะลดลง มีการลดลงของ B-lymphocytes ที่รับผิดชอบในการสังเคราะห์ IgA การเชื่อมโยง phagocytic ของภูมิคุ้มกันจะถูกละเมิดส่วนใหญ่เนื่องจากการละเมิด chemotaxis และการย่อยอาหารภายในเซลล์ใน neutrophils
ในการศึกษาการตรวจชิ้นเนื้อไตในผู้ป่วยที่มีอาการ Alport โดยกล้องจุลทรรศน์อิเล็กตรอนการเปลี่ยนแปลง ultrastructural สังเกตพังผืดไตชั้นใต้ดิน: ผอมบางและรูปแบบการแยกการละเมิดไตเยื่อชั้นใต้ดินที่มีการเปลี่ยนแปลงในความหนาและรูปทรงที่ไม่สม่ำเสมอของตน ในระยะแรกของโรคไตอักเสบทางพันธุกรรมข้อบกพร่องกำหนดความผอมบางและความเปราะบางของเยื่อหุ้มสมองกลวง
การเยื่อบาง ๆ ของเยื่อหุ้มปอดเป็นสัญญาณที่ดีมากและพบมากในเด็กหญิง คุณลักษณะกล้องจุลทรรศน์อิเล็กตรอนที่มีความคงที่มากกว่าในการเป็นโรคไตอักเสบทางพันธุกรรมคือความแตกแยกของเมมเบรนฐานและความรุนแรงของการทำลายของมันสัมพันธ์กับความรุนแรงของกระบวนการ

อาการของ Alport Syndrome ในเด็ก
อาการแรกของ Alport syndrome ในรูปแบบของโรคปัสสาวะที่แยกได้จะพบได้บ่อยในเด็ก ๆ ในช่วง 3 ปีแรกของชีวิต ในกรณีส่วนใหญ่โรคจะถูกตรวจพบโดยบังเอิญ กลุ่มอาการของทางเดินปัสสาวะจะถูกเปิดเผยในระหว่างการตรวจร่างกายเพื่อป้องกันเด็กก่อนเข้าสถาบันเด็กหรือในระหว่างการรักษาด้วยยา ARVI ในกรณีที่มีลักษณะทางพยาธิวิทยาในปัสสาวะระหว่าง ARVI ในโรคไตอักเสบทางพันธุกรรมซึ่งแตกต่างจากโรคไตวายเนื้องอกที่ได้รับไม่มีระยะแฝงอยู่
ในระยะเริ่มแรกของโรคความเป็นอยู่ที่ดีของเด็ก ๆ ได้รับความทุกข์ทรมานน้อยลักษณะเฉพาะคือความคงอยู่และความคงอยู่ของโรคทางเดินปัสสาวะ หนึ่งในสัญญาณหลักคือ hematuria ขององศาที่แตกต่างกันสังเกตเห็นใน 100% ของกรณี การเพิ่มขึ้นของระดับโลหิตเป็นเนื้องอกในระหว่างหรือหลังการติดเชื้อทางเดินหายใจการออกแรงกายหรือหลังการฉีดวัคซีนป้องกัน ภาวะโปรตีนในกรณีส่วนใหญ่ไม่เกิน 1 กรัมต่อวันเมื่อเริ่มมีอาการของโรคอาจจะไม่เสถียรเนื่องจากกระบวนการนี้ทำให้โปรตีนชักเพิ่มขึ้น เป็นระยะตะกอนปัสสาวะอาจมีเม็ดเลือดขาวที่มีความเด่นกว่าของ lymphocytes ซึ่งเกี่ยวข้องกับการพัฒนาการเปลี่ยนแปลงของคั่นระหว่างกัน
ต่อมามีการละเมิดการทำงานบางส่วนของไตอาการแย่ลงจากสภาพทั่วไปของผู้ป่วย: มึนเมากล้ามเนื้ออ่อนแอความดันเลือดแดงในหลอดเลือดแดงมักทำให้เกิดความบกพร่องในการได้ยิน (โดยเฉพาะในเด็กผู้ชาย) บางครั้งการมองเห็นบกพร่อง ความรู้สึกไม่สบายแสดงออกเป็นความรู้สึกอ่อนล้าปวดศีรษะ ในระยะแรกของโรคการสูญเสียการได้ยินในกรณีส่วนใหญ่จะตรวจพบโดยการบันทึกภาพเท่านั้น การสูญเสียการได้ยินใน Alport syndrome อาจเกิดขึ้นได้ในช่วงวัยเด็กที่แตกต่างกัน แต่ส่วนใหญ่แล้วการสูญเสียการได้ยินจะได้รับการวินิจฉัยเมื่ออายุ 6-10 ปี การสูญเสียการได้ยินในเด็กเริ่มต้นที่ความถี่สูงถึงระดับมากในอากาศและกระดูกการนำผ่านจากการดำเนินการเสียงเพื่อรับเสียงหูตึง การสูญเสียการได้ยินอาจเป็นหนึ่งในอาการแรกของโรคและสามารถนำไปสู่ภาวะปัสสาวะได้
ใน 20% ของกรณีผู้ป่วยที่มีอาการ Alport มีการเปลี่ยนแปลงในสายตา ความผิดปกติที่พบได้บ่อยที่สุดจากเลนส์: spherofokiya, lentikonus ล่วงหน้า, หลังหรือผสม, ความหลากหลายของต้อกระจก ในครอบครัวที่มีอาการ Alport มีอุบัติการณ์สำคัญของสายตาสั้น นักวิจัยหลายคนในครอบครัวเหล่านี้ทราบว่าการเปลี่ยนแปลงในระดับทวิภาคีในรูปของเม็ดสีขาวหรือสีเหลืองสดใสในบริเวณที่เป็นสีเหลือง พวกเขาถือว่าอาการนี้เป็นอาการคงที่ซึ่งมีค่าการวินิจฉัยสูงใน Alport syndrome C. S. Chugh et al. (1993) สำหรับการศึกษาตาเปิดเผย Alport ผู้ป่วยกลุ่มอาการของโรคลดลงในสายตา 66.7% ของกรณีไปข้างหน้า lenticonus - 37.8% จุดบนจอ - ใน 22,2%, ต้อกระจก - 20% keratoconus - 6 , 7%
ในเด็กบางคนที่มีโรคไตอักเสบทางพันธุกรรมโดยเฉพาะอย่างยิ่งในการก่อตัวของความไม่เพียงพอของไตความล้าหลังที่สำคัญในการพัฒนาทางกายภาพมีการระบุไว้ เนื่องจากความคืบหน้าของความไม่เพียงพอของไตจะพัฒนาความดันโลหิตสูง ในเด็กมักพบในวัยวัยรุ่นและในกลุ่มอายุมากขึ้น
ลักษณะคือการปรากฏตัวของผู้ป่วยโรคประสาทที่ถ่ายทอดทางพันธุกรรมของ stigmas ต่างๆ (มากกว่า 5-7) ในการเป็น dysembryogenesis ของเนื้อเยื่อเกี่ยวพัน ท่ามกลางเนื้อเยื่อเกี่ยวพันของความอัปยศในผู้ป่วยที่มี hypertelorism ตาที่พบบ่อยที่สุดเพดานสูงสบฟันรูปร่างที่ผิดปกติของหูโค้งของนิ้วก้อยในมือของเขา "sandalevidnaya ช่องว่าง" ที่เท้า สำหรับโรคไตอักเสบทางพันธุกรรมที่โดดเด่นด้วยความสม่ำเสมอ dizembriogeneza มลทินภายในครอบครัวเช่นเดียวกับความถี่สูงของการกระจายของพวกเขาในญาติ probands ผ่านที่โรคจะถูกส่ง
ในระยะแรกของโรคเผยให้เห็นการลดลงที่แยกจากการทำงานของไตบางส่วน: การขนส่งของกรดอะมิโนอิเล็กโทรฟังก์ชั่นความเข้มข้น Acidogenesis การเปลี่ยนแปลงต่อไปเป็นของรัฐการทำงานของทั้งสองใกล้ชิดและ nephron ปลายและมีลักษณะของความผิดปกติบางส่วนรวม การลดการกรองของไตจะเกิดขึ้นในเวลาต่อมาบ่อยขึ้นในช่วงวัยรุ่น เมื่อโรคไตวายเรื้อรังมีพัฒนาการจะเกิดภาวะโลหิตจางขึ้น
ดังนั้นสำหรับการถ่ายทอดทางพันธุกรรมโรคไตอักเสบการแสดงละครที่โดดเด่นของโรค: เวทีแฝงแรกหรือมีอาการทางคลินิกซ่อนประจักษ์โดยกลุ่มอาการของโรคกระเพาะปัสสาวะการเปลี่ยนแปลงน้อยที่สุดที่เกิดขึ้นแล้วกระบวนการ decompensation ค่อยเป็นค่อยไปกับการลดการทำงานของไตที่มีอาการทางคลินิกแจ่มแจ้ง (มัวเมาอ่อนแรงพัฒนาการล่าช้า anemizatsiya บริการ) อาการทางคลินิกปรากฏขึ้นโดยไม่คำนึงถึงการแบ่งชั้นของปฏิกิริยาการอักเสบ
โรคไตอักเสบทางพันธุกรรมสามารถปรากฏในช่วงอายุที่แตกต่างกันซึ่งขึ้นอยู่กับการกระทำของยีนซึ่งจนกว่าจะถึงเวลาที่กำหนดอยู่ในสถานะที่ถูกคุมขัง
การจัดหมวดหมู่
มีสามชนิดของโรคไตอักเสบทางพันธุกรรม
- I - มีอาการทางคลินิกด้วยโรคไตอักเสบที่มีก้อนเลือด, การสูญเสียการได้ยินและความเสียหายดวงตา โรคไตอักเสบเป็นพัฒนาการของ CRF ชนิดของการสืบทอดเป็นสิ่งสำคัญซึ่งเชื่อมโยงกับโครโมโซม X ในทางสัณฐานวิทยามีการรบกวนโครงสร้างของเมมเบรนพื้นฐานความผอมบางและความแตกแยก
- II เป็นอาการทางคลินิกด้วยโรคไตอักเสบที่มีก้อนเลือดออกโดยไม่สูญเสียการได้ยิน โรคไตอักเสบเป็นโรคไตวายเรื้อรังมากขึ้นเรื่อย ๆ ชนิดของการสืบทอดเป็นสิ่งสำคัญซึ่งเชื่อมโยงกับโครโมโซม X พบว่ามีการลดความบางลงของเยื่อบุผิวที่เป็นเส้นผ่านศูนย์กลาง (โดยเฉพาะลามินาเซีย)
- ตัวเลือกที่สาม - เลือดออกในครอบครัวที่ไม่เป็นอันตราย หลักสูตรเป็นที่ดีไตวายเรื้อรังไม่พัฒนา ชนิดของมรดกคือ autosomal dominant หรือ autosomal recessive ในประเภทของการถดถอย autosomal ผู้หญิงมักมีโรคร้ายแรงกว่า
การวินิจฉัยโรค Alport
เสนอเกณฑ์ต่อไปนี้:
- การมีอยู่ในครอบครัวอย่างน้อยสองรายที่เป็นโรคไต
- hematuria เป็นอาการชั้นนำของโรคไตใน proband;
- อย่างน้อยหนึ่งสมาชิกในครอบครัวมีการสูญเสียการได้ยิน;
- พัฒนาการไตวายเรื้อรังในญาติคนหนึ่งและอื่น ๆ
ในการวินิจฉัยของความหลากหลายของโรคทางพันธุกรรมและพิการ แต่กำเนิดเป็นสถานที่สำคัญเป็นวิธีการแบบบูรณาการเพื่อการตรวจสอบและเหนือสิ่งอื่นใดให้ความสำคัญกับข้อมูลที่ได้ในการจัดทำสายเลือดของเด็ก การวินิจฉัยกลุ่มอาการอัลพอร์ตถือว่าถูกต้องในกรณีที่ผู้ป่วย 3 ใน 4 คุณสมบัติทั่วไป: การปรากฏตัวในปัสสาวะครอบครัวและไตวายเรื้อรัง, การปรากฏตัวของการสูญเสียการได้ยินของผู้ป่วยประสาทที่โรคของการตรวจสอบในอิเล็กตรอนสัญญาณกล้องจุลทรรศน์ลักษณะการตรวจชิ้นเนื้อแตกแยกเยื่อฐานไตกับการเปลี่ยนแปลงของความหนาของมัน และรูปทรงที่ไม่สม่ำเสมอ
การตรวจสอบผู้ป่วยควรรวมถึงวิธีการตรวจทางพันธุกรรมทางคลินิก ชี้นำการศึกษาเกี่ยวกับการเกิดโรค; การตรวจโดยทั่วไปของผู้ป่วยโดยคำนึงถึงเกณฑ์การวินิจฉัยของบัญชี ขั้นตอนการชดเชยพยาธิวิทยาสามารถจับเพียงมุ่งเน้นไปที่กลุ่มอาการของโรคเช่นมีประวัติครอบครัว, ความดันโลหิตต่ำ, stigmas หลาย dizembriogeneza เปลี่ยนแปลงกลุ่มอาการของโรคกระเพาะปัสสาวะ ใน estrarenalnyh decompensated อาจทำให้เกิดอาการเช่นพิษรุนแรงอาการอ่อนเปลี้ยเพลียแรง, ปัญญาอ่อน anemizatsiya การพัฒนาทางกายภาพที่ประจักษ์และปรากฏการณ์ที่มีการลดลงอย่างค่อยเป็นค่อยไปของการทำงานของไต ในผู้ป่วยส่วนใหญ่ที่มีการลดลงของการทำงานของไตลดลงในการทำงานของ acido- และ aminogenesis สังเกต; ใน 50% ของผู้ป่วยสังเกตเห็นการลดลงอย่างมีนัยสำคัญในการทำงานของสารคัดหลั่งของไต; จำกัด ช่วงของความผันผวนของความหนาแน่นของปัสสาวะ; การละเมิดจังหวะของการกรองแล้วลดลงในการกรองไต ขั้นไตวายเรื้อรังได้รับการวินิจฉัยโดยการปรากฏตัวในผู้ป่วย 3-6 เดือนหรือระดับสูงมากขึ้นของยูเรียในซีรั่ม (มากกว่า 0.35 g / l) ลดลงกรองไตได้ถึง 25% ของปกติ
การวินิจฉัยที่แตกต่างกันของโรคไตอักเสบทางพันธุกรรมจะต้องดำเนินการโดยส่วนใหญ่จะเป็นรูปแบบของเลือดที่เป็นก้อนเลือดที่ได้รับ ได้รับ glomerulonephritis เฉียบพลันมากขึ้นเริ่มต้นระยะเวลา 2-3 สัปดาห์หลังจากการติดเชื้อก่อนหน้านี้คุณสมบัติ extrarenal รวมทั้งความดันโลหิตสูงกับวันแรก (ในโรคไตอักเสบพันธุกรรมตรงกันข้ามความดันเลือดต่ำ) ลดลงอัตราการกรองของไตที่เริ่มมีอาการ, การละเมิดของฟังก์ชั่นท่อบางส่วนไม่มีในขณะที่ เช่นเดียวกับพันธุกรรมที่มีอยู่ พบภาวะไตวายเนื้องอกที่เกิดขึ้นกับภาวะเลือดออกในกระแสเลือดและโปรตีนมากขึ้นโดยมี ESR เพิ่มขึ้น การเปลี่ยนแปลงทั่วไปในลักษณะเมมเบรนของเยื่อหุ้มสมองในไตมีความสำคัญในการวินิจฉัย
วินิจฉัยแยกโรคไต dysmetabolic ดำเนินการกับไตวายเรื้อรังที่อยู่ในครอบครัวที่ระบุคลินิกโรคไต Monotype และอาจมีตั้งแต่โรคไต pyelonephritis เพื่อ urolithiasis เด็ก ๆ มักมีอาการปวดท้องและปัสสาวะเป็นระยะ ๆ ในตะกอนปัสสาวะ - ออกซาเลต
หากคุณสงสัยว่าผู้ป่วยโรคไตอักเสบทางพันธุกรรมควรถูกส่งเพื่อชี้แจงการวินิจฉัยในแผนกโรคไตเฉพาะทาง
สิ่งที่ต้องตรวจสอบ?
วิธีการตรวจสอบ?
ต้องการทดสอบอะไรบ้าง?
ใครจะติดต่อได้บ้าง?
การรักษาโรค Alport
ในระบอบการปกครองให้สำหรับการ จำกัด การออกกำลังกายขนาดใหญ่อยู่ในที่มีอากาศบริสุทธิ์ อาหารที่มีคุณภาพสูงมีเนื้อหาโปรตีนและไขมันสูงไขมันและคาร์โบไฮเดรตเพียงพอโดยคำนึงถึงการทำงานของไต สิ่งสำคัญคือการระบุและการฟื้นฟูสมรรถภาพของโรคติดเชื้อเรื้อรัง จากยาเสพติด ATP, cocarboxylase, pyridoxine (ไม่เกิน 50 มก. / วัน) ใช้ carnitine chloride หลักสูตรนี้จัดขึ้น 2-3 ครั้งต่อปี เมื่อ hematuria มีการกำหนด phytotherapy - ตำแย, ตำแย, เถ้า BlackBerry, ยาร์โรว์
ในวรรณคดีต่างประเทศและในประเทศมีรายงานการรักษาด้วย prednisolone และการใช้ cytostatics อย่างไรก็ตามผลกระทบเป็นเรื่องยากที่จะตัดสิน
ในไตวายเรื้อรังล้มเหลว hemodialysis และปลูกถ่ายไต
ไม่มีวิธีการเฉพาะ (มีประสิทธิภาพ pathogenetic) บำบัดโรคไตอักเสบได้รับการถ่ายทอดทางพันธุกรรม มาตรการทางการแพทย์ทั้งหมดมีวัตถุประสงค์เพื่อป้องกันและชะลอการลดการทำงานของไต
อาหารควรมีความสมดุลและมีแคลอรีสูงโดยคำนึงถึงสถานะการทำงานของไต ในกรณีที่ไม่มีการละเมิดของรัฐในการทำงานของโภชนาการของเด็กควรจะมีเนื้อหาเพียงพอของโปรตีนไขมันและคาร์โบไฮเดรต ในกรณีที่มีอาการผิดปกติของไตปริมาณโปรตีนคาร์โบไฮเดรตของแคลเซียมและฟอสฟอรัสควรมี จำกัด ซึ่งจะทำให้พัฒนาการไตวายเรื้อรังล่าช้า
ความเครียดทางร่างกายควร จำกัด เด็กควรที่จะละเว้นจากการเล่นกีฬา
หลีกเลี่ยงการสัมผัสกับผู้ป่วยที่ติดเชื้อลดความเสี่ยงต่อการติดเชื้อทางเดินหายใจเฉียบพลัน มีความจำเป็นต้องล้างคราบจุลินทรีย์ที่ติดเชื้อเรื้อรัง การฉีดวัคซีนป้องกันสำหรับเด็กที่เป็นโรคไตอักเสบทางพันธุกรรมจะไม่ได้รับการดำเนินการการฉีดวัคซีนเป็นไปได้เฉพาะตามข้อบ่งชี้ทางระบาดวิทยา
การรักษาด้วยฮอร์โมนและภูมิคุ้มกันในโรคไตอักเสบทางพันธุกรรมไม่ได้ผล มีสัญญาณบ่งชี้ถึงผลดีบางอย่าง (การลดลงของระดับโปรตีนและการชะลอตัวของโรค) ด้วยการใช้ cyclosporine A และ ACE ในระยะยาวเป็นเวลาหลายปี
ในการรักษาผู้ป่วยที่ใช้ยาเสพติดที่ช่วยเพิ่มการเผาผลาญอาหาร:
- pyridoxine - 2-3 มก. / กก. / วันใน 3 ครั้งแบ่งเป็นเวลา 4 สัปดาห์;
- kokarboksilaza - 50 มก. ฉีดยาทุกวันอื่น ๆ เพียง 10-15 เม็ด;
- ATP - ฉีดเข้ากล้ามเนื้อทุกวัน 10 มิลลิลิตร
- วิตามิน A - 1000 U / ปี / วันใน 1 แผนกต้อนรับเป็นเวลา 2 สัปดาห์;
- วิตามินอี - 1 มก. / กก. / วันในแผนกต้อนรับ 1 ครั้งเป็นเวลา 2 สัปดาห์
การบำบัดดังกล่าวช่วยปรับปรุงสภาพทั่วไปของผู้ป่วยลดความผิดปกติของท่อและบริหารงานได้ 3 ครั้งต่อปี
Immunomodulator อาจใช้ levamisole - 2 มก. / กก. / วัน 2-3 ครั้งต่อสัปดาห์โดยมี intermissions ระหว่าง 3-4 วัน
นักวิจัยพบว่าการออกซิเจนในห้องปฏิบัติการ hyperbaric มีผลต่อความรุนแรงของภาวะเลือดออกและความผิดปกติของไต
วิธีที่มีประสิทธิภาพที่สุดในการรักษาโรคไตอักเสบทางพันธุกรรมคือการปลูกถ่ายไตในเวลาที่เหมาะสม เมื่อเป็นเช่นนี้จะไม่ได้สังเกตในการกำเริบของโรคปลูกในร้อยละขนาดเล็ก (ประมาณ 5%) อาจไตอักเสบการพัฒนาในการปลูกถ่ายไตที่เกี่ยวข้องกับแอนติเจนในเยื่อชั้นใต้ดินไต
พื้นที่ที่มีแนวโน้มคือการวินิจฉัยก่อนคลอดและการบำบัดทางพันธุกรรม การทดลองกับสัตว์แสดงให้เห็นถึงประสิทธิภาพในการถ่ายทอดยีนปกติที่มีความรับผิดชอบในการสังเคราะห์คอลลาเจนของสายพันธุกรรมชนิดที่หนึ่งไปยังเนื้อเยื่อไตหลังจากที่ได้มีการสังเคราะห์โครงสร้างคอลลาเจนตามปกติ
ภาพ
การพยากรณ์โรคของโรคไตอักเสบทางพันธุกรรมเป็นเรื่องร้ายแรงเสมอไป
เกณฑ์ที่ไม่เอื้ออำนวยต่อการไหลของโรคไตอักเสบทางพันธุกรรมคือ
- เพศชาย;
- พัฒนาการของไตวายเรื้อรังในครอบครัว
- proteinuria (มากกว่า 1 กรัม / วัน);
- ความหนาของเยื่อหุ้มสมองกลวงตามกล้องจุลทรรศน์;
- โรคประสาทอักเสบของเส้นประสาทหู;
- การลบยีน Co4A5
การพยากรณ์โรคของโลหิตเป็นปัสสาวะของครอบครัวที่ไม่เป็นอันตรายจะดีกว่า
Использованная литература