ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม (กลุ่มอาการอัลพอร์ต) ในเด็ก
ตรวจสอบล่าสุด: 05.07.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม (กลุ่มอาการอัลพอร์ต) เป็นโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมที่ไม่ใช่ภูมิคุ้มกัน โดยมีอาการแสดงคือมีเลือดในปัสสาวะ (บางครั้งมีโปรตีนในปัสสาวะ) การทำงานของไตลดลงอย่างต่อเนื่องจนกลายเป็นไตวายเรื้อรัง มักมีอาการหูหนวกจากการรับความรู้สึกทางประสาทและความบกพร่องทางการมองเห็นร่วมด้วย
โรคนี้ได้รับการอธิบายครั้งแรกในปี 1902 โดย LG Guthrie ซึ่งสังเกตครอบครัวหนึ่งซึ่งพบเลือดออกในปัสสาวะหลายชั่วอายุคน ในปี 1915 AF Hurst ได้อธิบายการพัฒนาของภาวะยูรีเมียในสมาชิกในครอบครัวเดียวกัน ในปี 1927 A. Alport ได้ระบุการสูญเสียการได้ยินในญาติหลายคนที่มีเลือดออกในปัสสาวะเป็นครั้งแรก ในช่วงทศวรรษปี 1950 ได้มีการอธิบายเกี่ยวกับรอยโรคที่ตาในโรคที่คล้ายกัน ในปี 1972 ในผู้ป่วยที่มีเลือดออกในปัสสาวะทางพันธุกรรม ในระหว่างการศึกษาสัณฐานวิทยาของเนื้อเยื่อไต Hinglais และคณะ พบว่ามีการขยายตัวและการแบ่งชั้นที่ไม่สม่ำเสมอของเยื่อฐานของไต ในปี 1985 ได้มีการระบุพื้นฐานทางพันธุกรรมของโรคไตอักเสบทางพันธุกรรม ซึ่งก็คือการกลายพันธุ์ในยีนคอลลาเจนชนิดที่ 4 (Fiengold et al., 1985)
การศึกษาลักษณะทางพันธุกรรมของโรคทำให้เราสรุปได้ว่าความแตกต่างในอาการแสดงทางฟีโนไทป์ของโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม (มีหรือไม่มีการสูญเสียการได้ยิน) เกิดจากระดับการแสดงออกของยีนกลายพันธุ์ ดังนั้น ในปัจจุบัน อาการทางคลินิกทั้งหมดถือเป็นอาการแสดงของโรคหนึ่งโรค และคำว่า "โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม" เป็นคำพ้องความหมายกับคำว่า "กลุ่มอาการอัลพอร์ต"
จากการศึกษาทางระบาดวิทยา พบว่าโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมเกิดขึ้นในอัตรา 17 ต่อเด็ก 100,000 คน
 [
[ สาเหตุของโรคอัลพอร์ต
พื้นฐานทางพันธุกรรมของโรคนี้คือการกลายพันธุ์ในยีนของคอลลาเจนชนิดที่ 4 ของโซ่ a-5 คอลลาเจนชนิดนี้พบได้ทั่วไปในเยื่อฐานของไต หูชั้นใน แคปซูลเลนส์ จอประสาทตา และกระจกตาของตา ซึ่งได้รับการพิสูจน์แล้วจากการศึกษาวิจัยที่ใช้แอนติบอดีโมโนโคลนอลต่อเศษส่วนคอลลาเจนนี้ เมื่อไม่นานมานี้ ได้มีการระบุถึงความเป็นไปได้ในการใช้โพรบ DNA เพื่อวินิจฉัยโรคไตอักเสบทางพันธุกรรมก่อนคลอด
ความสำคัญของการทดสอบสมาชิกในครอบครัวทั้งหมดด้วยโพรบ DNA เพื่อระบุพาหะของยีนกลายพันธุ์นั้นได้รับการเน้นย้ำ ซึ่งมีความสำคัญอย่างยิ่งในการให้คำปรึกษาทางการแพทย์และทางพันธุกรรมของครอบครัวที่มีโรคนี้ อย่างไรก็ตาม ครอบครัวมากถึง 20% ไม่มีญาติที่ป่วยเป็นโรคไต ซึ่งบ่งชี้ว่ามีการกลายพันธุ์ตามธรรมชาติของยีนที่ผิดปกติบ่อยครั้ง ผู้ป่วยโรคไตอักเสบทางพันธุกรรมส่วนใหญ่มีบุคคลในครอบครัวที่เป็นโรคไต สูญเสียการได้ยิน และมีปัญหาทางการมองเห็น การแต่งงานทางสายเลือดระหว่างผู้ที่มีบรรพบุรุษหนึ่งคนหรือมากกว่านั้นมีความสำคัญ เนื่องจากการแต่งงานของบุคคลที่เกี่ยวข้องกันนั้น โอกาสที่จะได้รับยีนเดียวกันจากทั้งพ่อและแม่จะเพิ่มขึ้น เส้นทางการถ่ายทอดทางพันธุกรรมที่ถ่ายทอดทางโครโมโซม X แบบเด่น ถ่ายทอดทางโครโมโซม X แบบด้อย และเด่น ได้รับการกำหนดขึ้นแล้ว
ในเด็ก โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมมักแบ่งได้เป็น 3 ประเภท คือ โรคอัลพอร์ต โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมโดยไม่สูญเสียการได้ยิน และเลือดออกในปัสสาวะชนิดไม่ร้ายแรงทางพันธุกรรม
โรคอัลพอร์ตเป็นโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมและมีความบกพร่องทางการได้ยิน เกิดจากความผิดปกติของโครงสร้างคอลลาเจนของเยื่อฐานของไต หู และตา ยีนของโรคอัลพอร์ตแบบคลาสสิกจะอยู่ที่ตำแหน่ง 21-22q ของแขนยาวของโครโมโซม X ในกรณีส่วนใหญ่ ยีนนี้จะถ่ายทอดทางพันธุกรรมแบบเด่น โดยเชื่อมโยงกับโครโมโซม X ในเรื่องนี้ โรคอัลพอร์ตจะรุนแรงกว่าในผู้ชาย เนื่องจากในผู้หญิง การทำงานของยีนกลายพันธุ์จะได้รับการชดเชยด้วยอัลลีลที่มีสุขภาพดีของโครโมโซมที่สองที่ไม่ได้รับความเสียหาย
พื้นฐานทางพันธุกรรมสำหรับการพัฒนาของโรคไตอักเสบทางพันธุกรรมคือการกลายพันธุ์ในยีนของโซ่แอลฟาของคอลลาเจนประเภท IV โซ่แอลฟาของคอลลาเจน G ประเภท IV 6 เป็นที่ทราบกัน: ยีนของโซ่ a5 และ a6 (Col4A5 และ Col4A5) อยู่บนแขนยาวของโครโมโซม X ในเขต 21-22q; ยีนของโซ่ a3 และ a4 (Col4A3 และ Col4A4) อยู่บนโครโมโซมที่ 2; ยีนของโซ่ a1 และ a2 (Col4A1 และ Col4A2) อยู่บนโครโมโซมที่ 13
ในกรณีส่วนใหญ่ (80-85%) จะตรวจพบรูปแบบการถ่ายทอดทางพันธุกรรมแบบ X-linked ของโรค ซึ่งเกี่ยวข้องกับความเสียหายของยีน Col4A5 อันเป็นผลจากการลบ การกลายพันธุ์แบบจุด หรือความผิดปกติของการต่อกัน ปัจจุบันพบการกลายพันธุ์ของยีน Col4A5 มากกว่า 200 กรณี ซึ่งส่งผลต่อการหยุดชะงักของการสังเคราะห์โซ่ a5 ของคอลลาเจนชนิดที่ 4 โรคนี้มักแสดงอาการในเด็กทั้งสองเพศ แต่ในเด็กผู้ชายจะมีอาการรุนแรงกว่า
การกลายพันธุ์ในโลคัสของยีน Col4A3 และ Col4A4 ที่รับผิดชอบในการสังเคราะห์โซ่ a3 และ a4 ของคอลลาเจนชนิดที่ 4 ถ่ายทอดทางออโตโซม ตามการวิจัยพบว่าการถ่ายทอดทางออโตโซมแบบเด่นพบได้ในผู้ป่วยโรคไตอักเสบที่ถ่ายทอดทางกรรมพันธุ์ 16% และแบบออโตโซมแบบด้อยพบได้ในผู้ป่วย 6% ยีน Col4A3 และ Col4A4 กลายพันธุ์ได้ประมาณ 10 รูปแบบ
ผลจากการกลายพันธุ์คือการละเมิดกระบวนการประกอบของคอลลาเจนประเภทที่ 4 ส่งผลให้โครงสร้างเสียหาย คอลลาเจนประเภทที่ 4 เป็นส่วนประกอบหลักอย่างหนึ่งของเยื่อฐานของไต อุปกรณ์หูชั้นใน และเลนส์ของตา ซึ่งพยาธิสภาพดังกล่าวจะถูกตรวจพบในคลินิกโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม
คอลลาเจนชนิดที่ 4 ซึ่งเป็นส่วนหนึ่งของเยื่อฐานของไต ประกอบด้วยโซ่ a1 (IV) สองโซ่และโซ่ a2 (IV) หนึ่งโซ่ และยังมีโซ่ a3, a4, a5 ด้วย โดยส่วนใหญ่แล้ว ในการถ่ายทอดทางพันธุกรรมแบบ X-link การกลายพันธุ์ของยีน Col4A5 มักจะมาพร้อมกับการขาดโซ่ a3, a4, a5 และ a6 ในโครงสร้างของคอลลาเจนชนิดที่ 4 และจำนวนโซ่ o1 และ a2 ในเยื่อฐานของไตเพิ่มขึ้น กลไกของปรากฏการณ์นี้ยังไม่ชัดเจน สันนิษฐานว่าสาเหตุคือการเปลี่ยนแปลงหลังการถอดรหัสใน mRNA
การไม่มีโซ่ a3, a4 และ a5 ในโครงสร้างของคอลลาเจนชนิดที่ 4 ของเยื่อฐานของไตทำให้บางลงและเปราะบางในระยะเริ่มแรกของโรคอัลพอร์ต ซึ่งอาการทางคลินิกมักแสดงออกมาด้วยภาวะเลือดออกในปัสสาวะ (มักแสดงออกมาน้อยกว่าด้วยภาวะเลือดออกในปัสสาวะร่วมกับโปรตีนในปัสสาวะหรือโปรตีนในปัสสาวะเพียงอย่างเดียว) สูญเสียการได้ยิน และเลนติโคนัส หากโรคดำเนินไปมากขึ้น โรคจะหนาขึ้นและซึมผ่านได้น้อยลงในระยะท้ายของโรค โดยมีคอลลาเจนชนิดที่ 5 และ 6 ขยายตัวขึ้น แสดงให้เห็นด้วยโปรตีนในปัสสาวะเพิ่มขึ้นและการทำงานของไตลดลง
ลักษณะของการกลายพันธุ์ที่อยู่เบื้องหลังโรคไตอักเสบทางพันธุกรรมนั้นกำหนดลักษณะทางฟีโนไทป์ของโรคเป็นส่วนใหญ่ ในกรณีของการขาดหายไปของโครโมโซม X พร้อมกับการกลายพันธุ์ของยีน Col4A5 และ Col4A6 ที่ทำหน้าที่ในการสังเคราะห์โซ่ a5 และ a6 ของคอลลาเจนชนิดที่ 4 กลุ่มอาการอัลพอร์ตจะรวมกับโรคกล้ามเนื้อเรียบของหลอดอาหารและอวัยวะสืบพันธุ์ ตามข้อมูลการวิจัย ในกรณีของการกลายพันธุ์ของยีน Col4A5 ที่เกี่ยวข้องกับการขาดหายไป จะสังเกตเห็นความรุนแรงของกระบวนการทางพยาธิวิทยาที่มากขึ้น โดยมีการรวมกันของความเสียหายของไตกับอาการภายนอกไตและการพัฒนาของไตวายเรื้อรังในระยะเริ่มต้น เมื่อเทียบกับการกลายพันธุ์แบบจุดของยีนนี้
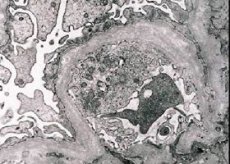
จากการศึกษาทางสัณฐานวิทยา กล้องจุลทรรศน์อิเล็กตรอนจะเผยให้เห็นการบางลงและการแบ่งชั้นของเยื่อฐานของไต (โดยเฉพาะ lamina densa) และการมีอยู่ของเม็ดเลือดที่มีอิเล็กตรอนหนาแน่น โรคไตอาจมีลักษณะแตกต่างกันในผู้ป่วยรายเดียวกัน ตั้งแต่โรคเมแซนเจียลเฉพาะจุดเล็กน้อยไปจนถึงโรคไตแข็ง โรคไตอักเสบในกลุ่มอาการอัลพอร์ตมักจะไม่มีผลทางภูมิคุ้มกัน ซึ่งทำให้แตกต่างจากโรคไตอักเสบ ลักษณะเด่น ได้แก่ การฝ่อของท่อไต การแทรกซึมของลิมโฟไซต์ และการมี "เซลล์โฟม" ที่มีการรวมตัวของไขมัน - ลิโปฟาจ เมื่อโรคดำเนินไป จะเผยให้เห็นการหนาขึ้นและการทำลายล้างที่ชัดเจนของเยื่อฐานของไต
การเปลี่ยนแปลงบางอย่างในระบบภูมิคุ้มกันถูกเปิดเผย ผู้ป่วยโรคไตอักเสบทางพันธุกรรมจะมีระดับ Ig A ลดลงและมีแนวโน้มที่จะมีความเข้มข้นของ IgM ในเลือดเพิ่มขึ้น โดยระดับ IgG อาจเพิ่มขึ้นในระยะเริ่มต้นของโรคและลดลงในระยะหลังๆ อาจเป็นไปได้ว่าการเพิ่มขึ้นของความเข้มข้นของ IgM และ G อาจเป็นปฏิกิริยาชดเชยชนิดหนึ่งที่ตอบสนองต่อภาวะขาด IgA
กิจกรรมการทำงานของระบบทีลิมโฟไซต์ลดลง สังเกตเห็นการลดลงอย่างเลือกสรรของบีลิมโฟไซต์ที่รับผิดชอบในการสังเคราะห์ Ig A การเชื่อมโยงการจับกินของภูมิคุ้มกันถูกขัดขวาง ซึ่งส่วนใหญ่เกิดจากการหยุดชะงักของกระบวนการเคมีแท็กซ์และการย่อยภายในเซลล์ในนิวโทรฟิล
เมื่อทำการตรวจชิ้นเนื้อไตในผู้ป่วยที่เป็นโรคอัลพอร์ต ข้อมูลจากกล้องจุลทรรศน์อิเล็กตรอนจะเผยให้เห็นการเปลี่ยนแปลงทางจุลภาคโครงสร้างของเยื่อฐานไต ได้แก่ การบางลง การทำลายโครงสร้าง และการแตกของเยื่อฐานไต โดยมีการเปลี่ยนแปลงความหนาและรูปร่างที่ไม่สม่ำเสมอ ในระยะเริ่มแรกของโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม ความผิดปกติจะกำหนดความบางและความเปราะบางของเยื่อฐานไต
การบางลงของเยื่อไตเป็นสัญญาณที่ดีกว่าและพบได้บ่อยในเด็กผู้หญิง สัญญาณที่เห็นได้ชัดในโรคไตอักเสบทางพันธุกรรมคือ การแตกของเยื่อฐาน และความรุนแรงของการทำลายเยื่อฐานจะสัมพันธ์กับความรุนแรงของกระบวนการนี้
อาการของโรคอัลพอร์ตในเด็ก
อาการเริ่มแรกของโรคอัลพอร์ตในรูปแบบของกลุ่มอาการทางเดินปัสสาวะแยกส่วนมักตรวจพบในเด็กอายุ 3 ปีแรกของชีวิต ในกรณีส่วนใหญ่ โรคนี้ตรวจพบโดยบังเอิญ กลุ่มอาการทางเดินปัสสาวะตรวจพบระหว่างการตรวจร่างกายเด็กก่อนเข้ารับการดูแลในสถานรับเลี้ยงเด็กหรือระหว่างการใช้ยาต้านไวรัสเอชไอวี ในกรณีที่มีพยาธิสภาพในปัสสาวะระหว่างการใช้ยาต้านไวรัสเอชไอวี ในโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม ซึ่งแตกต่างจากโรคไตอักเสบเรื้อรังที่เกิดขึ้นภายหลัง โรคนี้ไม่มีระยะแฝง
ในระยะเริ่มแรกของโรค สุขภาพของเด็กจะได้รับผลกระทบเพียงเล็กน้อย ลักษณะเด่นคืออาการปัสสาวะลำบากและดื้อยา อาการหลักอย่างหนึ่งคือปัสสาวะมีเลือดปนในระดับความรุนแรงที่แตกต่างกัน ซึ่งพบได้ 100% ของกรณี การเพิ่มขึ้นของระดับปัสสาวะมีขึ้นในระหว่างหรือหลังการติดเชื้อทางเดินหายใจ การออกกำลังกาย หรือหลังการฉีดวัคซีนป้องกัน โปรตีนในปัสสาวะในกรณีส่วนใหญ่ไม่เกิน 1 กรัมต่อวัน ในช่วงเริ่มต้นของโรคอาจไม่คงที่ เมื่อกระบวนการดำเนินไป โปรตีนในปัสสาวะจะเพิ่มขึ้น เป็นระยะๆ อาจมีเม็ดเลือดขาวในปัสสาวะที่มีลิมโฟไซต์เป็นส่วนใหญ่ในตะกอนของปัสสาวะ ซึ่งเกี่ยวข้องกับการพัฒนาของการเปลี่ยนแปลงในเนื้อเยื่อ
ต่อมา การทำงานของไตบางส่วนบกพร่อง อาการทั่วไปของผู้ป่วยจะแย่ลง มึนเมา กล้ามเนื้ออ่อนแรง ความดันโลหิตต่ำ มักสูญเสียการได้ยิน (โดยเฉพาะในเด็กผู้ชาย) และบางครั้งอาจสูญเสียการมองเห็น อาการมึนเมาจะแสดงออกมาเป็นสีซีด อ่อนล้า และปวดศีรษะ ในระยะเริ่มแรกของโรค การสูญเสียการได้ยินส่วนใหญ่จะตรวจพบได้จากการตรวจด้วยเครื่องตรวจเสียงเท่านั้น การสูญเสียการได้ยินในกลุ่มอาการอัลพอร์ตอาจเกิดขึ้นในช่วงต่างๆ ของวัยเด็ก แต่ส่วนใหญ่มักจะได้รับการวินิจฉัยเมื่ออายุ 6-10 ปี การสูญเสียการได้ยินในเด็กเริ่มจากความถี่สูง โดยไปถึงระดับที่สำคัญในอากาศและการนำเสียงทางกระดูก จากนั้นเปลี่ยนจากการสูญเสียการได้ยินจากการนำเสียงเป็นการสูญเสียการได้ยินจากการรับรู้เสียง การสูญเสียการได้ยินอาจเป็นอาการแรกๆ ของโรคและอาจเกิดขึ้นก่อนกลุ่มอาการทางเดินปัสสาวะ
ใน 20% ของกรณี ผู้ป่วยโรค Alport มีการเปลี่ยนแปลงของอวัยวะการมองเห็น ความผิดปกติที่ตรวจพบบ่อยที่สุดคือของเลนส์ ได้แก่ spherophokia, anterior, posterior หรือ mixed lenticonus และต้อกระจกต่างๆ ในครอบครัวที่มีโรค Alport มักมีภาวะสายตาสั้นจำนวนมาก นักวิจัยหลายคนสังเกตเห็นการเปลี่ยนแปลงของเยื่อบุตาทั้งสองข้างในครอบครัวเหล่านี้ในรูปแบบของเม็ดสีขาวสว่างหรือสีเหลืองใน corpus luteum พวกเขาถือว่าสัญญาณนี้เป็นอาการคงที่ที่มีค่าสูงในการวินิจฉัยโรค Alport KS Chugh et al. (1993) จากการศึกษาจักษุวิทยาพบว่าในผู้ป่วยโรค Alport การมองเห็นลดลงใน 66.7% ของกรณี anterior lenticonus ใน 37.8% จุดบนจอประสาทตาใน 22.2% ต้อกระจกใน 20% และกระจกตาโป่งพองใน 6.7%
ในเด็กบางคนที่มีโรคไตอักเสบทางพันธุกรรม โดยเฉพาะเมื่อไตวายเกิดขึ้น การพัฒนาทางร่างกายจะล่าช้าอย่างมาก เมื่อไตวายดำเนินไป ความดันโลหิตสูงก็จะค่อยๆ พัฒนาขึ้น ในเด็ก มักตรวจพบในช่วงวัยรุ่นและกลุ่มอายุที่มากขึ้น
ผู้ป่วยโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมมีลักษณะเด่นคือมีตราประทับของเนื้อเยื่อเกี่ยวพันผิดปกติ (มากกว่า 5-7 ตราประทับ) ในบรรดาตราประทับของเนื้อเยื่อเกี่ยวพันในผู้ป่วย ตราประทับที่พบบ่อยที่สุด ได้แก่ ตาเหล่เกินปกติ เพดานปากสูง ความผิดปกติของการสบฟัน รูปร่างผิดปกติของใบหู นิ้วก้อยของมือโค้งงอ และ "ช่องว่างระหว่างนิ้วเท้า" โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมมีลักษณะเด่นคือตราประทับของเนื้อเยื่อเกี่ยวพันผิดปกติที่สม่ำเสมอกันภายในครอบครัวเดียวกัน รวมถึงมีการกระจายของตราประทับเหล่านี้บ่อยครั้งในญาติของผู้ที่เป็นโรคนี้
ในระยะเริ่มแรกของโรค จะตรวจพบการลดลงของการทำงานของไตบางส่วน ได้แก่ การขนส่งกรดอะมิโน อิเล็กโทรไลต์ การทำงานของความเข้มข้น การสร้างกรด การเปลี่ยนแปลงในภายหลังจะส่งผลต่อสถานะการทำงานของส่วนต้นและส่วนปลายของหน่วยไต และมีลักษณะเฉพาะคือความผิดปกติบางส่วนร่วมกัน การลดลงของการกรองของไตจะเกิดขึ้นในภายหลัง โดยมักเกิดขึ้นในช่วงวัยรุ่น เมื่อไตอักเสบทางพันธุกรรมดำเนินไป จะเกิดภาวะโลหิตจาง
ดังนั้น โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมจึงมีลักษณะเฉพาะคือมีการดำเนินโรคเป็นขั้นตอน ขั้นแรกคืออาการทางคลินิกที่แฝงอยู่หรืออาการทางคลินิกที่แสดงออกโดยการเปลี่ยนแปลงเล็กน้อยในกลุ่มอาการทางเดินปัสสาวะ จากนั้นกระบวนการจะค่อยๆ เสื่อมลงโดยการทำงานของไตลดลงพร้อมกับอาการทางคลินิกที่ชัดเจน (มึนเมา อ่อนแรง พัฒนาการล่าช้า โลหิตจาง) อาการทางคลินิกมักจะปรากฏขึ้นโดยไม่คำนึงถึงปฏิกิริยาอักเสบที่เกิดขึ้น
โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมสามารถแสดงอาการได้ในช่วงอายุต่างๆ ซึ่งขึ้นอยู่กับการทำงานของยีนที่อยู่ในสถานะถูกกดไว้จนถึงช่วงเวลาหนึ่ง
การจำแนกประเภท
โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมมี 3 ชนิด
- ตัวเลือกที่ 1 - อาการทางคลินิกคือไตอักเสบร่วมกับมีเลือดในปัสสาวะ สูญเสียการได้ยิน และตาเสียหาย ไตอักเสบจะค่อยๆ ลุกลามขึ้นเรื่อยๆ จนกลายเป็นไตวายเรื้อรัง การถ่ายทอดทางพันธุกรรมนั้นมีความโดดเด่น โดยเชื่อมโยงกับโครโมโซม X จากลักษณะทางสัณฐานวิทยา พบว่ามีการละเมิดโครงสร้างของเยื่อฐาน ทำให้เยื่อฐานบางลงและแตกออก
- ตัวเลือกที่ 2 - อาการทางคลินิกคือไตอักเสบร่วมกับมีเลือดในปัสสาวะโดยไม่สูญเสียการได้ยิน อาการไตอักเสบจะค่อยๆ แย่ลงเมื่อไตวายเรื้อรังพัฒนาไป ประเภทของอาการถ่ายทอดเป็นแบบเด่นซึ่งเชื่อมโยงกับโครโมโซม X ในทางสัณฐานวิทยา ตรวจพบการบางลงของเยื่อฐานของหลอดเลือดฝอยในไต (โดยเฉพาะ laminadensa)
- ตัวเลือกที่ 3 - ภาวะเลือดออกในปัสสาวะในครอบครัวที่ไม่ร้ายแรง อาการค่อนข้างดี ไม่เกิดภาวะไตวายเรื้อรัง การถ่ายทอดทางพันธุกรรมเป็นแบบถ่ายทอดทางพันธุกรรมแบบ...
การวินิจฉัยโรคอัลพอร์ต
มีเกณฑ์เสนอดังนี้:
- การมีผู้ป่วยโรคไตในครอบครัวละอย่างน้อย 2 ราย
- ภาวะเลือดออกในปัสสาวะเป็นอาการหลักของโรคไตในผู้ป่วยทดสอบ
- การมีการสูญเสียการได้ยินในสมาชิกในครอบครัวอย่างน้อยหนึ่งคน
- การพัฒนาของภาวะไตวายเรื้อรังในญาติหนึ่งรายหรือมากกว่านั้น
ในการวินิจฉัยโรคทางพันธุกรรมและโรคประจำตัวต่างๆ มักจะให้ความสำคัญกับการตรวจวินิจฉัยอย่างครอบคลุม และที่สำคัญที่สุดคือต้องให้ความสำคัญกับข้อมูลที่ได้จากการรวบรวมข้อมูลประวัติครอบครัวของเด็ก การวินิจฉัยโรคอัลพอร์ตจะถือว่าถูกต้องในกรณีที่ตรวจพบอาการทั่วไป 3 ใน 4 อาการในผู้ป่วย ได้แก่ การมีเลือดออกในปัสสาวะและไตวายเรื้อรังในครอบครัว การสูญเสียการได้ยินจากประสาทรับความรู้สึก ความผิดปกติของการมองเห็นในผู้ป่วย การตรวจพบสัญญาณของการแยกตัวของเยื่อฐานของไตที่มีการเปลี่ยนแปลงความหนาและรูปร่างที่ไม่สม่ำเสมอระหว่างการตรวจชิ้นเนื้อด้วยกล้องจุลทรรศน์อิเล็กตรอน
การตรวจร่างกายผู้ป่วยควรใช้วิธีการวิจัยทางคลินิกและทางพันธุกรรม การศึกษาประวัติโรคอย่างมีเป้าหมาย การตรวจร่างกายทั่วไปของผู้ป่วยโดยคำนึงถึงเกณฑ์ที่สำคัญในการวินิจฉัย ในระยะชดเชย สามารถตรวจพบพยาธิวิทยาได้โดยเน้นที่กลุ่มอาการต่างๆ เช่น การมีภาระทางพันธุกรรม ความดันโลหิตต่ำ การเกิดตัวอ่อนผิดปกติหลายจุด การเปลี่ยนแปลงในกลุ่มอาการทางเดินปัสสาวะ ในระยะชดเชย อาจมีอาการนอกไต เช่น มึนเมาอย่างรุนแรง อ่อนแรง พัฒนาการทางร่างกายล่าช้า โลหิตจาง อาการเหล่านี้แสดงออกมาและรุนแรงขึ้นพร้อมกับการทำงานของไตที่ลดลงอย่างค่อยเป็นค่อยไป ในผู้ป่วยส่วนใหญ่ที่การทำงานของไตลดลง จะสังเกตได้ดังนี้ การสร้างกรดและกรดอะมิโนลดลง ผู้ป่วย 50% พบว่าการทำงานของไตลดลงอย่างมีนัยสำคัญ ความหนาแน่นของแสงในปัสสาวะมีความผันผวนในช่วงจำกัด จังหวะการกรองผิดปกติ และการทำงานของไตลดลง ระยะของไตวายเรื้อรังได้รับการวินิจฉัยเมื่อผู้ป่วยมีระดับยูเรียในซีรั่มเลือดสูง (มากกว่า 0.35 กรัมต่อลิตร) นาน 3-6 เดือนขึ้นไป และอัตราการกรองของไตลดลงเหลือ 25% ของค่าปกติ
การวินิจฉัยแยกโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมควรทำโดยหลักแล้วด้วยโรคไตอักเสบชนิดมีเลือดในปัสสาวะ โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมมักมีอาการเฉียบพลันในช่วง 2-3 สัปดาห์หลังจากการติดเชื้อ มีอาการภายนอกไต เช่น ความดันโลหิตสูงตั้งแต่วันแรก (ในทางตรงกันข้ามในโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม ความดันโลหิตต่ำ) การกรองของไตลดลงในช่วงเริ่มต้นของโรค ไม่มีการทำงานของท่อไตบางส่วนบกพร่อง ในขณะที่โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมจะมีอาการดังกล่าว โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมมักมีอาการเลือดออกในปัสสาวะและโปรตีนในปัสสาวะมากขึ้น โดยมีค่า ESR สูงขึ้น การเปลี่ยนแปลงทั่วไปของเยื่อฐานของไต ซึ่งเป็นลักษณะเฉพาะของโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม ถือเป็นประโยชน์ต่อการวินิจฉัย
การวินิจฉัยแยกโรคไตจากภาวะการเผาผลาญผิดปกติจะดำเนินการกับภาวะไตวายเรื้อรัง ในครอบครัวมีโรคไตที่แตกต่างกันทางคลินิก และอาจมีกลุ่มของโรคไตตั้งแต่ไตอักเสบจนถึงนิ่วในทางเดินปัสสาวะ เด็กมักบ่นว่าปวดท้อง และมีตะกอนในปัสสาวะเป็นระยะๆ เช่น ออกซาเลต
หากสงสัยว่าเป็นโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรม ควรส่งผู้ป่วยไปที่แผนกโรคไตเฉพาะทางเพื่อชี้แจงการวินิจฉัย
สิ่งที่ต้องตรวจสอบ?
วิธีการตรวจสอบ?
ต้องการทดสอบอะไรบ้าง?
ใครจะติดต่อได้บ้าง?
การรักษาอาการอัลพอร์ตซินโดรม
ระบอบการรักษานี้รวมถึงการจำกัดการออกกำลังกายอย่างหนักและการสัมผัสกับอากาศบริสุทธิ์ รับประทานอาหารให้ครบถ้วน มีโปรตีน ไขมัน และคาร์โบไฮเดรตในปริมาณที่เพียงพอ โดยคำนึงถึงการทำงานของไต สิ่งสำคัญอย่างยิ่งคือการตรวจหาและรักษาการติดเชื้อเรื้อรัง ยาที่ใช้ ได้แก่ ATP, cocarboxylase, pyridoxine (สูงสุด 50 มก./วัน), carnitine chloride รับประทาน 2-3 ครั้งต่อปี สำหรับภาวะเลือดออกในปัสสาวะ แพทย์จะสั่งยาสมุนไพร เช่น ตำแย น้ำคั้นจากต้นโช้กเบอร์รี่ และยาร์โรว์
มีรายงานในวรรณกรรมต่างประเทศและในประเทศเกี่ยวกับการรักษาด้วยเพรดนิโซโลนและการใช้ยาไซโตสแตติก อย่างไรก็ตาม ยากที่จะตัดสินผลได้
ในกรณีไตวายเรื้อรัง จะมีการฟอกไตและปลูกถ่ายไต
ยังไม่มีวิธีการบำบัดโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมโดยเฉพาะ (ได้ผล) มาตรการการรักษาทั้งหมดมุ่งเป้าไปที่การป้องกันและชะลอการเสื่อมถอยของการทำงานของไต
การรับประทานอาหารควรสมดุลและมีแคลอรีสูง โดยคำนึงถึงสภาพการทำงานของไต ในกรณีที่ไม่มีความผิดปกติทางการทำงาน อาหารของเด็กควรมีโปรตีน ไขมัน และคาร์โบไฮเดรตในปริมาณที่เพียงพอ ในกรณีที่มีอาการไตเสื่อม ควรจำกัดปริมาณโปรตีน คาร์โบไฮเดรต แคลเซียม และฟอสฟอรัส ซึ่งจะชะลอการเกิดภาวะไตวายเรื้อรัง
ควรจำกัดกิจกรรมทางกาย แนะนำให้เด็กๆ หลีกเลี่ยงการเล่นกีฬา
ควรหลีกเลี่ยงการสัมผัสกับผู้ป่วยติดเชื้อ ลดความเสี่ยงในการเกิดโรคทางเดินหายใจเฉียบพลัน จำเป็นต้องทำความสะอาดบริเวณที่ติดเชื้อเรื้อรัง ไม่ฉีดวัคซีนป้องกันสำหรับเด็กที่มีโรคไตอักเสบทางพันธุกรรม การฉีดวัคซีนทำได้เฉพาะเมื่อมีข้อบ่งชี้ทางระบาดวิทยาเท่านั้น
การบำบัดด้วยฮอร์โมนและยากดภูมิคุ้มกันในโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมไม่ได้ผล มีข้อบ่งชี้ถึงผลดีบางประการ (ปริมาณโปรตีนในปัสสาวะลดลงและโรคดำเนินไปช้าลง) จากการใช้ไซโคลสปอรินเอและสารยับยั้งเอซีอีเป็นระยะเวลานานหลายปี
ในการรักษาคนไข้จะมีการใช้ยาที่ช่วยเพิ่มการเผาผลาญ ได้แก่
- ไพริดอกซิน - 2-3 มก./กก./วัน แบ่งเป็น 3 ครั้ง เป็นเวลา 4 สัปดาห์
- โคคาร์บอกซิเลส - 50 มก. ฉีดเข้ากล้ามเนื้อ ทุกวันเว้นวัน รวม 10-15 ครั้ง
- ATP - ฉีดเข้ากล้าม 1 มล. ทุกวันเว้นวัน จำนวน 10-15 ครั้ง
- วิตามินเอ 1,000 IU/ปี/วัน แบ่งรับประทาน 1 ครั้ง เป็นเวลา 2 สัปดาห์
- วิตามินอี 1 มก./กก./วัน แบ่งรับประทาน 1 ครั้ง เป็นเวลา 2 สัปดาห์
การบำบัดประเภทนี้จะช่วยปรับปรุงสภาพทั่วไปของผู้ป่วย ลดความผิดปกติของหลอดไต และดำเนินการเป็นคอร์ส 3 ครั้งต่อปี
Levamisole สามารถใช้เป็นยาปรับภูมิคุ้มกันได้ 2 มก./กก./วัน สัปดาห์ละ 2-3 ครั้ง โดยเว้นระยะระหว่างการให้ยา 3-4 วัน
ตามข้อมูลการวิจัย พบว่าการให้ออกซิเจนภายใต้แรงดันสูงมีผลดีต่อความรุนแรงของภาวะเลือดออกในปัสสาวะและความผิดปกติของไต
วิธีการรักษาโรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมที่มีประสิทธิผลที่สุดคือการปลูกถ่ายไตให้ทันเวลา ในกรณีนี้ โรคจะไม่กำเริบอีกในระหว่างการปลูกถ่าย ในบางกรณี (ประมาณ 5%) อาจเกิดโรคไตอักเสบในไตที่ปลูกถ่ายซึ่งเกี่ยวข้องกับแอนติเจนที่เยื่อฐานของไต
แนวทางที่มีแนวโน้มดีคือการวินิจฉัยก่อนคลอดและการบำบัดด้วยวิศวกรรมพันธุกรรม การทดลองกับสัตว์แสดงให้เห็นประสิทธิภาพสูงในการถ่ายโอนยีนปกติที่รับผิดชอบในการสังเคราะห์โซ่คอลลาเจนอัลฟาชนิดที่ 4 เข้าไปในเนื้อเยื่อไต หลังจากนั้นจึงสังเกตการสังเคราะห์โครงสร้างคอลลาเจนปกติ
พยากรณ์
การพยากรณ์โรคไตอักเสบที่ถ่ายทอดทางพันธุกรรมมักเป็นเรื่องสำคัญเสมอ
เกณฑ์การพยากรณ์โรคที่ไม่พึงประสงค์สำหรับการดำเนินของโรคไตอักเสบทางพันธุกรรม ได้แก่:
- เพศชาย;
- การพัฒนาระยะเริ่มต้นของภาวะไตวายเรื้อรังในสมาชิกในครอบครัว
- โปรตีนในปัสสาวะ (มากกว่า 1 กรัม/วัน)
- ความหนาของเยื่อฐานของไตตามการตรวจด้วยกล้องจุลทรรศน์
- โรคเส้นประสาทอักเสบ
- การลบออกจากยีน Col4A5
การพยากรณ์โรคสำหรับภาวะเลือดออกในปัสสาวะแบบทางพันธุกรรมที่ไม่ร้ายแรงมีแนวโน้มดีขึ้น
Использованная литература