ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
Alcaptonuria - พยาธิวิทยาของเอนไซม์ที่มีมา แต่กำเนิด
ตรวจสอบล่าสุด: 23.04.2024

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
หนึ่งในความผิดปกติของการเผาผลาญที่หายากมาก - alkaptonuria - หมายถึงความผิดปกติ แต่กำเนิดในการเผาผลาญของกรดอะมิโนไทโรซีน
นอกจากนี้ โรคนี้สามารถเรียกได้ว่าขาด homogentisate oxidase, homogentisinuria, โรคทางพันธุกรรมหรือโรคปัสสาวะสีดำ [1]
ระบาดวิทยา
จากสถิติพบว่ามีอัลคัปโตนูเรียไม่เกิน 9 รายต่อประชากร 1 ล้านคน และในประเทศส่วนใหญ่ในยุโรป - หนึ่งกรณีต่อการเกิดมีชีพ 100-250,000 ราย
ในบรรดาประเทศในยุโรป ข้อยกเว้นคือสโลวาเกีย (โดยเฉพาะภูมิภาคที่ค่อนข้างเล็กทางตะวันตกเฉียงเหนือ) ซึ่งความชุกของอัลคัปโตนูเรียเป็นหนึ่งในกรณีของทารกแรกเกิด 19,000 คน เป็นไปได้ทั้งหมด เนื่องจากในครอบครัวของสโลวักโรมาที่อาศัยอยู่ที่นั่น ระดับการผสมพันธุ์ (การแต่งงานระหว่างลูกพี่ลูกน้อง) สูงที่สุดในยุโรป: 10-14% [2]
สาเหตุ alkaptonuria
สาเหตุที่แท้จริงของ alkaptonuria เนื่องจากความผิดปกติ แต่กำเนิดของ catabolism (การสลายตัวของเมตาบอลิซึม) ของอะโรมาติก (homocyclic) α-amino acid tyrosine ได้รับการจัดตั้งขึ้น: ความผิดปกติของการเผาผลาญนี้เป็นผลมาจากการกลายพันธุ์ของ homozygous หรือ heterozygous ที่ซับซ้อนจากหนึ่งในพัน ของยีนบนโครโมโซม 3 อย่างแม่นยำยิ่งขึ้น ยีน HqG21 ที่ locus 3 -q23 บนแขนยาวของโครโมโซม ยีนนี้เข้ารหัสลำดับนิวคลีโอไทด์ของเอนไซม์ตับ homogentisate-1,2-dioxygenase [3](เรียกอีกอย่างว่า homogentisic acid oxidase หรือ homogentisate oxidase) ซึ่งเป็นโลหะที่มีธาตุเหล็กซึ่งจำเป็นสำหรับขั้นตอนหนึ่งของการสลายตัวของไทโรซีนในร่างกาย [4], [5]
ดังนั้น alkaptonuria จึงเป็นข้อบกพร่องในเอนไซม์ homogentisate-1,2-dioxygenase ที่แม่นยำยิ่งขึ้นซึ่งเป็นผลมาจากความบกพร่องทางพันธุกรรมที่กำหนดหรือขาดหายไปอย่างสมบูรณ์ [6]
การเป็นภาวะหมักที่มีมา แต่กำเนิดนั้น alkaptonuria ได้รับการถ่ายทอดเป็นลักษณะถอย autosomal นั่นคือเพื่อให้ alkaptonuria เกิดขึ้นในเด็กพ่อแม่ทั้งสองต้องมียีนดัดแปลงสำหรับเอนไซม์นี้เนื่องจากแต่ละคนส่งสำเนาของเอนไซม์นี้ไปให้เด็กเพียงสำเนาเดียว จากยีนทั้งสองที่มีอยู่
ตามข้อมูลล่าสุด มีการดัดแปลงยีน HGD มากกว่าสองร้อยแบบ และมักพบการกลายพันธุ์ที่ผิดพลาด การโยกย้าย และการประกบกัน
ปัจจัยเสี่ยง
ปัจจัยเสี่ยงเพียงอย่างเดียวสำหรับการพัฒนาของ fermentopathy ที่มีมา แต่กำเนิดนี้คือประวัติครอบครัวและการสืบทอดของยีน HGD ที่ดัดแปลงสองชุดหากผู้ปกครองไม่แสดง alkaptonuria (ความเสี่ยงของการถ่ายทอดความผิดปกติคือ 25%) หรืออย่างใดอย่างหนึ่ง ผู้ปกครองมีความผิดปกตินี้ [7]
กลไกการเกิดโรค
ไทโรซีนมีบทบาทสำคัญในการสังเคราะห์โปรตีน การผลิตโครโมโปรตีน - เม็ดสีผิวเมลานิน ฮอร์โมนไทรอยด์ และสารสื่อประสาทคาเทโคลามีน
กลไกในการควบคุมปริมาณไทโรซีนในเซลล์นั้นซับซ้อนมาก และร่างกายจะทำให้เนื้อหาส่วนเกินเป็นปกติโดยการแยกส่วน กระบวนการแคแทบอลิซึมของไทโรซีน เช่นเดียวกับกรดอะมิโนอะโรมาติกทั้งหมด มีหลายขั้นตอนและดำเนินไปในหลายขั้นตอน นอกจากนี้แต่ละขั้นตอนของการสลายตัวเมตาบอลิซึมของไทโรซีนเกิดขึ้นด้วยการมีส่วนร่วมของเอนไซม์บางชนิดและการก่อตัวของสารประกอบระดับกลาง
อย่างแรกเลย กรดอะมิโนจะถูกแยกออกเป็นพารา-ไฮดรอกซีฟีนิลไพรูเวต ซึ่งจะกลายเป็นอัลคาโปน - 2,5-ไดไฮดรอกซีฟีนิลอะซีติกหรือกรดโฮโมเจนติซิก นอกจากนี้ อัลคาโปนควรเปลี่ยนเป็นกรดมาเลอะซิติก แต่สิ่งนี้ไม่เกิดขึ้น [8]
และการเกิดโรคของ alkaptonuria คือการหยุดปฏิกิริยาทางชีวเคมีของ tyrosine catabolism ในขั้นตอนของการก่อตัวของกรด homogentisic: ไม่มีเอนไซม์ที่จำเป็นในการทำลายมัน - homogentisate oxidase
ร่างกายไม่ได้ใช้กรด Homogentisic และสามารถสะสมด้วยการขับถ่ายทางไต นอกจากนี้ มันถูกออกซิไดซ์เป็นเบนโซควิโนอะซีเตต (กรดเบนโซควิโนนอะซิติก) ซึ่งเมื่อจับกับโมเลกุลของเนื้อเยื่อและของเหลวในร่างกาย จะเกิดสารประกอบไบโอโพลีเมอร์ที่มีสีเหมือนเมลานิน
การสะสมของผลิตภัณฑ์ขั้นกลางเหล่านี้ในเนื้อเยื่อนำไปสู่การหยุดชะงักของโครงสร้างคอลลาเจนของเนื้อเยื่อกระดูกอ่อนเนื่องจากความยืดหยุ่นลดลง - ด้วยอาการทางคลินิกหลายอย่างของ alkaptonuria และการพัฒนาของภาวะแทรกซ้อน
อาการ alkaptonuria
Alcaptonuria ในทารกแรกเกิดและทารกเป็นที่ประจักษ์โดยความมืดของปัสสาวะ เมื่อสัมผัสกับอากาศ สีของปัสสาวะบนผ้าอ้อม ผ้าอ้อม และชุดชั้นในจะเปลี่ยนเป็นสีน้ำตาลเข้ม นี่เป็นเพราะการสะสมและการปล่อยกรดโฮโมเจนติซิกซึ่งถูกออกซิไดซ์เป็นเบนโซควิโนอะซิเตต [9]
ในกรณีที่ไม่มีอาการอื่น ๆ ภาวะอัลคัปโตนูเรียในเด็กตั้งแต่อายุยังน้อยมักไม่เป็นที่รู้จักอย่างทันท่วงที เนื่องจากหลังจากปัสสาวะ ปัสสาวะอาจมืดลงหลังจากผ่านไปสองสามชั่วโมง ตามรายงานบางฉบับ ในเงื่อนไขของคลินิก ตรวจพบว่ามีเพียงหนึ่งในห้าของเด็กอายุต่ำกว่า 12 เดือนที่เกิดมาพร้อมกับโรคหมักดองนี้เท่านั้น ดังนั้นจึงเป็นสิ่งสำคัญมากสำหรับผู้ปกครองที่จะต้องใส่ใจในการดูแลทารก
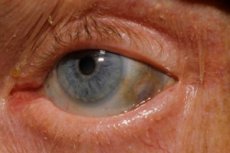
นอกจากนี้ สัญญาณเริ่มต้น ได้แก่ เม็ดสี (สีเทาอมฟ้า) ของตาขาวและกระดูกอ่อนของใบหูและจมูกซึ่งมักเรียกกันว่า ochronosis [10]
เมื่อเวลาผ่านไป อาการอื่น ๆ จะปรากฏขึ้น:
- ผิวคล้ำอย่างรุนแรงบนโหนกแก้มรักแร้และอวัยวะเพศ
- การย้อมเสื้อผ้าเมื่อสัมผัสกับส่วนที่มีเหงื่อออกของร่างกาย
- การโจมตีของความอ่อนแอทั่วไป
- เสียงแหบ.
ควรระลึกไว้เสมอว่า alkaptonuria และ ochronosis ตามที่ระบุไว้ข้างต้นเป็นชื่อที่มีความหมายเหมือนกันสำหรับการละเมิดแคแทบอลิซึมของไทโรซีนเช่นเดียวกัน
โรคน้ำเชื่อมเมเปิ้ลและอัลคัปโตนูเรีย โรคเมเปิลไซรัปที่มีมา แต่กำเนิดหรือลิวซิโนซิสยังหมายถึงความผิดปกติของการเผาผลาญ มีการสืบทอดประเภทเดียวกัน และแม้กระทั่งการกลายพันธุ์ก็เกิดขึ้นบนโครโมโซมเดียวกัน แต่เกี่ยวข้องกับยีนที่เข้ารหัสคอมเพล็กซ์ของเอนไซม์ของกรดดีไฮโดรจีเนสที่มีกิ่งก้าน ด้วยเหตุนี้ ร่างกายจึงไม่สามารถทำลายส่วนประกอบโปรตีนบางชนิดได้ โดยเฉพาะกรดอะมิโน ลิวซีน ไอโซลิวซีน และวาลีน ในภาวะนี้ ปัสสาวะ (และขี้หู) มีกลิ่นที่หอมหวาน นอกจากนี้ในภาพทางคลินิกของกรดอินทรีย์ชนิดนี้, hypopigmentation, ความผันผวนของความดันโลหิต, ชัก, อาเจียนและท้องร่วง, ระดับน้ำตาลในเลือดลดลง, ketoacidosis, ภาพหลอน ฯลฯ อัตราการเสียชีวิตในเด็กค่อนข้างมาก สูง; ในผู้ใหญ่ที่ไม่ได้รับการรักษา อาจโคม่าและเสียชีวิตได้เนื่องจากสมองบวมน้ำ
Albinism และ alkaptonuria "รวม" เฉพาะไทโรซีนเท่านั้น โรคเผือกรวมถึงโรคเผือกที่ตา เกิดจากการกลายพันธุ์ทางพันธุกรรมที่ส่งผลต่อการผลิตเม็ดสีเมลานิน การเปลี่ยนแปลงที่มีมาแต่กำเนิดพบในยีน TYR บนโครโมโซม 11 (11q14.3) ซึ่งเข้ารหัสไทโรซิเนส เอนไซม์ที่ประกอบด้วยทองแดงในเมลาโนโซมซึ่งจำเป็นสำหรับการสร้างเม็ดสีผิวโดยพิจารณาจากผลิตภัณฑ์เมตาบอลิซึมของไทโรซีน โรคนี้พบได้บ่อยกว่าอัลคัปโตนูเรียมาก
ภาวะแทรกซ้อนและผลกระทบ
ผลที่ตามมาและภาวะแทรกซ้อนของ alkaptonuria ที่เกิดจากการกระทำของสารไทโรซีนระดับกลาง - กรด homogentisic และ benzoquinoneacetic - ปรากฏขึ้นเนื่องจากการสะสมของโพลีเมอร์เม็ดสีปฏิกิริยาการทำลายเส้นใยคอลลาเจนและการเสื่อมสภาพของกระดูกอ่อน ต่อความเค้นทางกล)
หลายปีต่อมาในวัยผู้ใหญ่โรคข้อเสื่อมและโรคข้อเข่าเสื่อมของข้อต่อขนาดใหญ่ (สะโพก sacroiliac และเข่า) พัฒนา การหดตัวของช่องว่าง intervertebral (โดยเฉพาะกระดูกสันหลังส่วนเอวและทรวงอก) - ด้วยการกลายเป็นปูนและการก่อตัวของ osteophytes; ความหนาแน่นของเนื้อเยื่อของแผ่นกระดูก subchondral ลดลง และกระดูกที่อยู่เบื้องล่างสามารถรับการเปลี่ยนแปลงทางพยาธิวิทยาด้วยการก่อตัวของการเจริญเติบโตและการเสียรูป [11]
ความเสียหายต่อลิ้นหัวใจ (เอออร์ติกและไมตรัล) และหลอดเลือดหัวใจที่มีอาการของโรคหัวใจขาดเลือด เช่นเดียวกับการก่อตัวของนิ่วในไตและต่อมลูกหมาก เนื่องจากการกลายเป็นปูนเดียวกัน อาจเกิดขึ้น [12], [13]
การวินิจฉัย alkaptonuria
โดยปกติ การวินิจฉัยความผิดปกติของการเผาผลาญแต่กำเนิดจะขึ้นอยู่กับการศึกษาของเหลวในร่างกาย
บนพื้นฐานของการทดสอบใดและปฏิกิริยาใดที่สามารถวินิจฉัย alkaptonuria ได้? จำเป็นต้องมีการทดสอบปัสสาวะ - เพื่อตรวจหากรด homogentisic และกำหนดระดับ (ปกติ - 20-30 มก. ต่อวันเพิ่มขึ้น - 3-8 กรัม) ตัวอย่างปัสสาวะถูกตรวจสอบโดยแก๊สโครมาโตกราฟีหรือแมสสเปกโตรเมทรีโดยใช้โครมาโตกราฟีของเหลว การตรวจคัดกรองหาเฟอริกคลอไรด์ในปัสสาวะเป็นไปได้ [14]
นอกจากนี้ยังมีวิธีการวินิจฉัยอย่างรวดเร็ว - การกำหนดอัลแคปตันในจุดปัสสาวะแห้งบนกระดาษ (ตามความเข้มของสี)
เมื่อชี้แจงการวินิจฉัยโรค การวินิจฉัยด้วยเครื่องมือ (X-ray) เกี่ยวข้องกับการระบุสัญญาณเอ็กซ์เรย์ของโรคข้อเข่าเสื่อมและโรคข้ออื่น ๆ ในผู้ป่วย
การวินิจฉัยได้รับการยืนยันโดย& วิธีทางอณูพันธุศาสตร์ เช่น การวินิจฉัยโรคทางพันธุกรรมเช่น การทดสอบทางพันธุกรรมและการจัดลำดับดีเอ็นเอ [15]
การวินิจฉัยที่แตกต่างกัน
การวินิจฉัยแยกโรครวมถึง hemochromatosis และตับวายเฉียบพลันของทารกแรกเกิด, melaninuria, porphyria ไม่สม่ำเสมอเฉียบพลัน, lymphohistiocytosis เม็ดเลือด, พยาธิสภาพของยลหลัก, โรคไขข้ออักเสบ, ankylosing spondylitis
ใครจะติดต่อได้บ้าง?
การรักษา alkaptonuria
การรักษาหลักสำหรับ alkaptonuria คือการกลืนกินกรดแอสคอร์บิกในปริมาณมาก (อย่างน้อย 1,000 มก. ต่อวัน) ในเด็ก สิ่งนี้จะเพิ่มการขับกรดโฮโมเจนติซิกในปัสสาวะ และในผู้ใหญ่จะลดระดับอนุพันธ์ของกรดเบนโซควิโนนอะซิติกในปัสสาวะ และชะลอการยึดเกาะกับโครงสร้างเนื้อเยื่อเกี่ยวพันของข้อต่อและคอลลาเจน [16]
ในคลินิกในยุโรปตะวันตก ยา Nitizinone (Orfalin) กำลังได้รับการทดสอบ ซึ่งเป็นยาของกลุ่มสารเมตาโบไลต์ที่ยับยั้งขั้นตอนที่สองของแคแทบอลิซึมของไทโรซีน: การเปลี่ยนแปลงของพารา-ไฮดรอกซีฟีนิลไพรูเวตเป็นกรดโฮโมเจนติซิก อย่างไรก็ตาม การใช้สารทางเภสัชวิทยานี้ทำให้เกิดการสะสมของไทโรซีนและอาจทำให้เกิดผลข้างเคียงที่รุนแรงได้ เช่น ความทึบของกระจกตาและอาการกลัวแสง เลือดออกทางจมูกและในกระเพาะอาหาร ตับวาย การเปลี่ยนแปลงของเลือด เป็นต้น อย่างไรก็ตาม ในสหรัฐอเมริกา Nithizinone ได้รับการอนุมัติจากองค์การอาหารและยาสำหรับการรักษา ไทโรซินีเมีย ชนิดที่ 1 [17], [18]
ด้วยเหตุนี้ กายภาพบำบัด - การออกกำลังกายบำบัดเพื่อเพิ่มความแข็งแรงของกล้ามเนื้อและเพิ่มการเคลื่อนไหวของข้อต่อ balneo และ peloidotherapy เพื่อลดความเจ็บปวด - ดำเนินการสำหรับปัญหาข้อต่อที่เกิดจาก alkaptonuria
แม้ว่าไทโรซีนไม่ได้ให้มาแต่อาหารเท่านั้น แต่ยังผลิตในร่างกายด้วย ผู้ป่วยอัลคัปโตนูเรียควรรับประทานอาหารที่มีโปรตีนต่ำและจำกัดการบริโภคอาหารที่อุดมด้วยไทโรซีน โดยเฉพาะเนื้อวัวและเนื้อหมู ผลิตภัณฑ์จากนม (โดยเฉพาะชีส) พืชตระกูลถั่ว ถั่ว ฯลฯ เมล็ดพืช
การป้องกัน
การป้องกันการกลายพันธุ์ของยีนเป็นไปไม่ได้ และเพื่อป้องกันการเกิดของเด็กที่มีความเสี่ยงสูงต่อความผิดปกติแต่กำเนิด มีการให้คำปรึกษาทางการแพทย์และทางพันธุกรรมซึ่งจำเป็นก่อนการตั้งครรภ์ตามแผนของคู่สมรสที่มีประวัติครอบครัวเป็นโรคทางพันธุกรรม ธรรมชาติ. [19]
พยากรณ์
ภาวะอัลคัปโตนูเรียมีผู้เสียชีวิตน้อยมาก และการเสียชีวิตอาจเกิดจากโรคแทรกซ้อนร้ายแรงที่ส่งผลต่อหัวใจและไต ดังนั้นสำหรับอายุขัยโดยรวมของผู้ที่มี alkaptonuria การพยากรณ์โรคเป็นสิ่งที่ดี
แต่คุณภาพชีวิตลดลงเนื่องจากอาการปวดอย่างรุนแรงในข้อต่อหรือกระดูกสันหลัง โดยมีข้อ จำกัด ด้านการเคลื่อนไหวอย่างมากซึ่งมักจะก้าวหน้า