ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
โรคกระจกตา: สาเหตุ อาการ การวินิจฉัย การรักษา
ตรวจสอบล่าสุด: 07.07.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
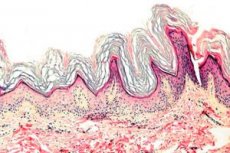
โรคผิวหนังชนิดหนึ่งที่มีลักษณะเฉพาะคือมีการสร้างเคราตินผิดปกติมากเกินไป โดยมักเกิดที่ฝ่ามือและฝ่าเท้า
สาเหตุและพยาธิสภาพของโรคยังไม่ชัดเจน การวิจัยได้พิสูจน์แล้วว่าโรคผิวหนังกระจกตาเกิดจากการกลายพันธุ์ของยีนที่เข้ารหัสเคราติน 6, 9, 16 การขาดวิตามินเอ ความผิดปกติของฮอร์โมน โดยเฉพาะต่อมเพศ การติดเชื้อแบคทีเรียและไวรัส ล้วนมีความสำคัญอย่างยิ่งต่อพยาธิสภาพของโรคนี้ ซึ่งเป็นอาการหนึ่งของโรคทางพันธุกรรมและเนื้องอกของอวัยวะภายใน (โรคผิวหนังกระจกตาแบบพาราปโซเรียติก)
อาการ แบ่งออกเป็นโรคกระจกตาชนิดกระจาย (Unna-Tost keratoderma, Meleda keratoderma, Papillon-Lefevre keratoderma, mutilating keratoderma และกลุ่มอาการที่รวมถึงโรคกระจกตาชนิดกระจายเป็นอาการหลักอย่างหนึ่ง) และโรคกระจกตาชนิดกระจายเฉพาะจุด (โรคกระจกตาที่มีจุดกระจายของ Fischer-Buschke, โรคกระจกตาแบบมีจุดกระจายของ Kosti, โรคกระจกตาแบบจำกัดของ Bruhauer-Franzesthesti, โรคกระจกตาแบบเส้นตรงของ Fuchs เป็นต้น)
โรคผิวหนังอักเสบ Winy-Tost (คำพ้องความหมาย: โรคผิวหนังอักเสบแต่กำเนิดที่ฝ่ามือและฝ่าเท้า, กลุ่มอาการ Winy-Tost) ถ่ายทอดทางพันธุกรรมแบบถ่ายทอดทางพันธุกรรม การเกิดเคราตินมากเกินไปที่ฝ่ามือและฝ่าเท้า (บางครั้งเกิดเฉพาะฝ่าเท้า) จะเกิดขึ้นในช่วง 2 ปีแรกของชีวิต กระบวนการทางพยาธิวิทยาของผิวหนังเริ่มต้นด้วยการที่ผิวหนังบริเวณฝ่ามือและฝ่าเท้าหนาขึ้นเล็กน้อยเป็นแถบสีแดงเข้มบริเวณขอบผิวหนังที่แข็งแรง เมื่อเวลาผ่านไป อาจมีชั้นขนสีเหลืองเรียบๆ ปรากฏขึ้นบนพื้นผิว รอยโรคนี้ไม่ค่อยลามไปถึงหลังข้อมือหรือหลังนิ้ว ในผู้ป่วยบางราย อาจมีรอยแตกร้าวที่ผิวเผินหรือลึก และพบภาวะเหงื่อออกมากเกินไปในบริเวณนั้น ในผู้ป่วยที่ผู้เขียนสังเกตพบ ลุงฝ่ายแม่ พี่ชาย และลูกชาย ป่วยเป็นโรคผิวหนังอักเสบ Winy-Tost
บรรยายกรณีความเสียหายของเล็บ (หนาขึ้น) ฟัน และผมในโรคผิวหนัง Winy-Tost keratoderma ร่วมกับความผิดปกติของโครงกระดูกต่างๆ และพยาธิสภาพของอวัยวะภายใน ระบบประสาท และระบบต่อมไร้ท่อ
การตรวจทางพยาธิวิทยา การตรวจทางพยาธิวิทยาพบภาวะผิวหนังหนาผิดปกติ มีเนื้อเยื่อหนา ผิวหนังหนา และมีการอักเสบเล็กน้อยในชั้นหนังแท้ส่วนบน การวินิจฉัยแยกโรค ต้องแยกโรคนี้จากโรคผิวหนังกระจกตาชนิดอื่น
โรคผิวหนังชนิด Meleda keratoderma (คำพ้องความหมาย: โรค Meleda, โรคผิวหนังชนิด acrokeratoma ที่ลุกลามแต่กำเนิด, โรคผิวหนังชนิด Siemens' palmoplantar transgradient keratosis, โรคผิวหนังชนิด Kogoy's hereditary palmoplantar progressive keratosis) ถ่ายทอดทางพันธุกรรมแบบถ่ายทอดทางยีนลักษณะด้อย โรคผิวหนังชนิดนี้มีลักษณะเป็นชั้นหนาสีเหลืองน้ำตาลที่มีรอยแตกลึก ขอบของรอยโรคเป็นสีม่วงอมม่วงกว้างหลายมิลลิเมตร โดยทั่วไปแล้วกระบวนการนี้จะลามไปที่หลังมือและเท้า ปลายแขน และหน้าแข้ง ผู้ป่วยส่วนใหญ่จะมีภาวะเหงื่อออกมากเกินไปในบริเวณนั้น ในกรณีนี้ ผิวของฝ่ามือและฝ่าเท้าจะชื้นเล็กน้อยและมีจุดสีดำปกคลุม (ท่อต่อมเหงื่อ)
โรคนี้สามารถเกิดขึ้นได้ในช่วงอายุ 15-20 ปี เล็บจะหนาขึ้นและผิดรูป
การตรวจทางพยาธิวิทยา การตรวจทางพยาธิวิทยาพบภาวะผิวหนังหนาผิดปกติ บางครั้งมีภาวะผิวหนังหนา และการอักเสบเรื้อรังในชั้นหนังแท้
การวินิจฉัยแยกโรค ต้องแยกโรคผิวหนังชนิด Melela keratoderma จากโรคผิวหนังชนิด Unna-Tost keratoderma
Keratoderma Papillon-Lefevre (คำพ้องความหมาย: โรคผิวหนังหนาบริเวณฝ่ามือและฝ่าเท้าร่วมกับโรคปริทันต์อักเสบ) ถ่ายทอดทางพันธุกรรมแบบถ่ายทอดทางยีนลักษณะด้อย
โรคนี้จะแสดงอาการในช่วงปีที่ 2-3 ของชีวิต ภาพทางคลินิกของโรคนี้คล้ายกับโรคเมเลลา นอกจากนี้ การเปลี่ยนแปลงของฟันก็มีลักษณะเฉพาะเช่นกัน (ความผิดปกติของการขึ้นของฟันน้ำนมและฟันแท้พร้อมกับการเกิดฟันผุ โรคเหงือกอักเสบ โรคปริทันต์ที่ลุกลามอย่างรวดเร็วพร้อมกับการสูญเสียฟันก่อนวัยอันควร)
การตรวจทางพยาธิวิทยา การตรวจทางพยาธิวิทยาพบการหนาขึ้นของชั้นต่างๆ ของหนังกำพร้า โดยเฉพาะชั้นที่มีขน และกลุ่มเซลล์ลิมโฟไซต์และฮิสทิโอไซต์ในชั้นหนังแท้มีจำนวนน้อย
การวินิจฉัยแยกโรค ควรแยกโรคนี้จากโรคกระจกตาชนิดอื่น ๆ ลักษณะเด่นที่สำคัญคือพยาธิสภาพของฟันที่มีลักษณะเฉพาะซึ่งไม่พบในโรคกระจกตาชนิดแพร่กระจายที่ถ่ายทอดทางพันธุกรรมชนิดอื่น
Keratoderma mutilans (คำพ้องความหมาย: Fonwinkel syndrome, hereditary mutilating keratoma) เป็นโรคผิวหนังชนิดหนึ่งที่ถ่ายทอดทางพันธุกรรมแบบถ่ายทอดทางพันธุกรรมแบบออโตโซมเด่น โดยจะพัฒนาขึ้นในปีที่ 2 ของชีวิต และมีลักษณะเฉพาะคือมีคราบไขมันเกาะอยู่ทั่วผิวหนังบริเวณฝ่ามือและฝ่าเท้า ซึ่งทำให้เกิดภาวะเหงื่อออกมากเกินปกติ เมื่อเวลาผ่านไป รอยหยักคล้ายเส้นเอ็นจะก่อตัวขึ้นที่นิ้วมือ ซึ่งนำไปสู่การหดเกร็งและการตัดนิ้วมือโดยธรรมชาติ การเกิด keratosis ของรูขุมขนจะเกิดขึ้นที่หลังมือ รวมถึงบริเวณข้อศอกและข้อเข่า แผ่นเล็บจะเปลี่ยนแปลง (มักคล้ายกับแว่นตา) มีรายงานกรณีของภาวะฮอร์โมนเพศชายต่ำ ผมร่วงเป็นหย่อม สูญเสียการได้ยิน และโรคแพคิโอนีเชีย
การตรวจทางพยาธิวิทยา การตรวจทางพยาธิวิทยาพบภาวะผิวหนังหนาผิดปกติอย่างรุนแรง มีเนื้อเยื่อเป็นก้อนหนา ผิวหนังหนา และมีการอักเสบเล็กน้อยในชั้นหนังแท้ ซึ่งประกอบด้วยลิมโฟไซต์และฮิสทิโอไซต์
การวินิจฉัยแยกโรค เมื่อแยกโรคกระจกตาอักเสบชนิดทำลายเนื้อเยื่อจากโรคกระจกตาอักเสบชนิดแพร่กระจายชนิดอื่น ควรคำนึงถึงผลกระทบจากการทำลายเนื้อเยื่อก่อนเป็นอันดับแรก ซึ่งไม่ใช่ลักษณะทั่วไปของโรคกระจกตาอักเสบชนิดอื่น เมื่อทำการวินิจฉัยแยกโรคกระจกตาอักเสบชนิดแพร่กระจายทุกรูปแบบ จำเป็นต้องจำไว้ว่าโรคนี้อาจเป็นอาการหลักอย่างหนึ่งของโรคทางพันธุกรรมหลายชนิด
การรักษา Neotigazone ระบุไว้ในการรักษาโรคผิวหนังกระจกตาโดยทั่วไป ปริมาณยาขึ้นอยู่กับความรุนแรงของกระบวนการและอยู่ที่ 0.3-1 มก./กก. ของน้ำหนักผู้ป่วย ในกรณีที่ไม่มี Neotigazone แนะนำให้ใช้วิตามินเอในปริมาณ 100 ถึง 300,000 มก. ต่อวันเป็นเวลานาน การบำบัดภายนอกประกอบด้วยการใช้ยาทาที่มีเรตินอยด์อะโรมาติก สารที่ทำลายกระจกตา และสารสเตียรอยด์
 [
[ สิ่งที่รบกวนคุณ?
สิ่งที่ต้องตรวจสอบ?
วิธีการตรวจสอบ?