สิ่งตีพิมพ์ใหม่
ภาวะศีรษะสามหัว
ตรวจสอบล่าสุด: 04.07.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
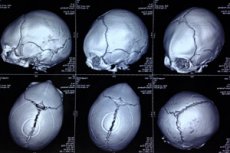
ความผิดปกติแต่กำเนิดในรูปแบบของความผิดปกติของกะโหลกศีรษะ ซึ่งศีรษะของทารกมีรูปร่างไม่สม่ำเสมอและกะโหลกศีรษะปรากฏเป็นสามเหลี่ยม เรียกว่า trigonocephaly (จากภาษากรีก trigonon – สามเหลี่ยม และ kephale – ศีรษะ) [ 1 ]
ระบาดวิทยา
อัตราการเกิดภาวะกะโหลกศีรษะปิดไม่สนิทอยู่ที่ประมาณ 5 รายต่อทารกเกิดมีชีวิต 10,000 ราย (หรือ 1 รายต่อประชากร 2,000–2,500 คนในประชากรทั่วไป) [ 2 ]
ใน 85% ของกรณี ภาวะกะโหลกศีรษะปิดไม่สนิทเกิดขึ้นโดยไม่สม่ำเสมอ ส่วนที่เหลือจะเกิดขึ้นเป็นส่วนหนึ่งของกลุ่มอาการ [ 3 ]
ตามสถิติ การหลอมรวมของรอยต่อหน้าผากส่วนกลางก่อนกำหนดเป็นรูปแบบที่พบบ่อยเป็นอันดับสองของภาวะกะโหลกศีรษะปิด และภาวะศีรษะสามหัวพบได้ 1 กรณีในทารกแรกเกิด 5,000-15,000 ราย จำนวนทารกเพศชายที่มีความผิดปกตินี้เกือบสามเท่าของทารกเพศหญิง [ 4 ]
ในประมาณ 5% ของกรณี ความผิดปกติแต่กำเนิดนี้ปรากฏอยู่ในประวัติครอบครัว [ 5 ]
สาเหตุ ภาวะศีรษะสามหัว
การสร้างกะโหลกศีรษะให้ปกติเกิดขึ้นเนื่องจากมีศูนย์การเจริญเติบโตขั้นต้นและการสร้างกระดูกใหม่ – ข้อต่อระหว่างกะโหลกศีรษะและใบหน้า ซึ่งจะปิดลงในช่วงเวลาหนึ่งระหว่างการพัฒนาของโครงกระดูกศีรษะเพื่อให้แน่ใจว่ากระดูกจะเชื่อมติดกัน [ 6 ]
กระดูกหน้าผาก (os frontale) ของกะโหลกศีรษะของทารกแรกเกิดประกอบด้วยสองส่วน โดยมีการเชื่อมต่อแบบเส้นใยแนวตั้งระหว่างสองส่วน นั่นคือ รอยประสานหน้าผากส่วนกลางหรือรอยประสานเมโทปิก (จากภาษากรีก metopon ซึ่งแปลว่าหน้าผาก) ซึ่งทอดยาวจากด้านบนของสันจมูกขึ้นไปตามแนวกลางของหน้าผากจนถึงกระหม่อมด้านหน้า รอยประสานกะโหลกศีรษะแบบเส้นใยนี้เป็นเพียงรอยประสานเดียวเท่านั้นที่จะสมานตัวในวัยทารก ตั้งแต่อายุ 3-4 เดือนจนถึงอายุ 8-18 ปี [ 7 ]
ดูเพิ่มเติม – การเปลี่ยนแปลงของกะโหลกศีรษะหลังคลอด
สาเหตุของภาวะศีรษะสามหัว ได้แก่ภาวะกะโหลก ศีรษะปิดไม่สนิท (craniosynostosis) หรือภาวะศีรษะสามหัว (metopic synostosis) (จากภาษากรีก syn – รวมกัน และ osteon – กระดูก) นั่นคือ การหลอมรวมของกระดูกของกะโหลกศีรษะตามรอยต่อระหว่างหน้าผากส่วนกลางก่อนกำหนด (ก่อนอายุ 3 เดือน) ดังนั้น ภาวะกะโหลกศีรษะปิดไม่สนิทและภาวะศีรษะสามหัวจึงเกี่ยวข้องกันในฐานะสาเหตุและผล หรือเป็นกระบวนการทางพยาธิวิทยาและผลลัพธ์ [ 8 ]
ในกรณีส่วนใหญ่ ภาวะศีรษะสามหัวในเด็กเป็นผลมาจากภาวะกะโหลกศีรษะปิดไม่สนิทแบบแยกส่วน (Craniosynostosis) ซึ่งสาเหตุที่แน่ชัดยังไม่ทราบ ภาวะกะโหลกศีรษะปิดไม่สนิทแบบแยกส่วนเกิดขึ้นเป็นครั้งคราว ซึ่งอาจเกิดจากปัจจัยทางพันธุกรรมและสิ่งแวดล้อมร่วมกัน [ 9 ]
แต่ภาวะศีรษะสามหัวอาจเป็นส่วนหนึ่งของกลุ่มอาการแต่กำเนิดที่เกิดจากความผิดปกติของโครโมโซมและการกลายพันธุ์ของยีนต่างๆ ได้แก่ กลุ่มอาการศีรษะสามหัวของออปิตซ์ (กลุ่มอาการโบห์ริง-ออปิตซ์), กลุ่มอาการอาเพิร์ต, กลุ่มอาการโลยส์-ดิเอตซ์, กลุ่มอาการไฟฟ์เฟอร์, กลุ่มอาการแจ็กสัน-ไวส์, กลุ่มอาการผิดปกติของกะโหลกศีรษะและใบหน้า หรือกลุ่มอาการครูซอน, กลุ่มอาการจาคอบเซน, กลุ่มอาการแซทเทรอ-โชตเซน และกลุ่มอาการมุนเคอ ในกรณีดังกล่าว ภาวะศีรษะสามหัวจะเรียกว่ากลุ่มอาการ [ 10 ]
เมื่อแรกเกิด สมองจะมีขนาดประมาณ 25% ของขนาดผู้ใหญ่ และเมื่อสิ้นสุดปีแรกของชีวิต สมองจะมีขนาดประมาณ 75% ของสมองผู้ใหญ่ แต่ด้วยความล่าช้าในการเจริญเติบโตของสมองในขั้นต้น อาจเกิดภาวะที่เรียกว่า craniosynostosis ขั้นที่สองได้ สาเหตุของความล่าช้านี้เกี่ยวข้องกับความผิดปกติของระบบเผาผลาญ โรคทางโลหิตวิทยาบางชนิด และผลของสารเคมีที่ก่อให้เกิดความพิการแต่กำเนิดต่อทารกในครรภ์ (รวมถึงสารเคมีในผลิตภัณฑ์ยา) [ 11 ]
ตามที่ผู้เชี่ยวชาญระบุว่า ภาวะศีรษะสามหัวในผู้ใหญ่ที่ไม่ได้รับการรักษาในวัยเด็กอันเป็นผลจากภาวะกะโหลกศีรษะปิดไม่สนิทหรืออาการพิการแต่กำเนิดจะคงอยู่ตลอดชีวิต [ 12 ]
ปัจจัยเสี่ยง
ผู้เชี่ยวชาญเชื่อว่าปัจจัยเสี่ยงหลักที่ทำให้เกิดภาวะศีรษะสามหัว (และภาวะกะโหลกศีรษะปิดไม่สนิทก่อนวัยเป็นสาเหตุ) เป็นเรื่องทางพันธุกรรม ในช่วงสองทศวรรษที่ผ่านมา มีการระบุยีนได้มากกว่า 60 ยีน ซึ่งการกลายพันธุ์มีความเกี่ยวข้องกับการหลอมรวมของกระดูกกะโหลกศีรษะก่อนวัยอันควรและไม่สามารถเคลื่อนไหวได้ในทารก
ความเสี่ยงในการเกิดโรคข้อกระดูกและกระดูกใบหน้าและกะโหลกศีรษะอักเสบและการสร้างกระดูกโดยทั่วไปจะเพิ่มขึ้นในกรณีที่ทารกมีรูปร่างผิดปกติ ภาวะขาดออกซิเจนในมดลูก การตั้งครรภ์แฝด การดื่มแอลกอฮอล์ การใช้ยา หรือการสูบบุหรี่ในระหว่างตั้งครรภ์ [ 13 ]
กลไกการเกิดโรค
ตามทฤษฎีที่เป็นที่ยอมรับ พยาธิสภาพของภาวะศีรษะสามหัวมีสาเหตุมาจากการหยุดชะงักของการสร้างกระดูกของทารกในครรภ์ในระยะแรกของการตั้งครรภ์ ซึ่งส่วนใหญ่มักเกิดจากปัจจัยทางพันธุกรรม เนื่องจากตรวจพบความผิดปกติของโครโมโซมแบบสุ่มในทารกแรกเกิดที่มีภาวะกะโหลกศีรษะปิดไม่สนิท ตัวอย่างเช่น ภาวะที่พบบ่อยที่สุดอย่างหนึ่งคือภาวะไตรโซมี 9p ซึ่งนำไปสู่ข้อบกพร่องของกะโหลกศีรษะ ใบหน้า และโครงกระดูก ปัญญาอ่อน และพัฒนาการทางจิตพลศาสตร์ [ 14 ]
เนื่องมาจากการเชื่อมรอยต่อระหว่างกระดูกหน้าผากส่วนกลางเร็วเกินไป การเจริญเติบโตในบริเวณนี้ของกะโหลกศีรษะจึงถูกขัดขวาง การเจริญเติบโตด้านข้างของกระดูกหน้าผากจะถูกจำกัดด้วยการสั้นลงของโพรงกะโหลกศีรษะด้านหน้า สันกระดูกจะก่อตัวขึ้นตามแนวกึ่งกลางของหน้าผาก มีกระดูกที่ก่อตัวเป็นเบ้าตามาบรรจบกันและรอยบุ๋มของกระดูกขมับ [ 15 ]
แต่การเจริญเติบโตของกะโหลกศีรษะในบริเวณอื่น ๆ ยังคงดำเนินต่อไป: มีการเจริญเติบโตชดเชยในแนวซากิตตัล (ด้านหน้าไปด้านหลัง) และแนวขวางของด้านหลังของกะโหลกศีรษะ (พร้อมกับการขยายตัวของส่วนข้างขม่อม-ท้ายทอย) เช่นเดียวกับการเจริญเติบโตในแนวตั้งและแนวซากิตตัลของส่วนบนของใบหน้า อันเป็นผลมาจากความผิดปกติเหล่านี้ กะโหลกศีรษะจึงมีรูปร่างไม่สม่ำเสมอ - เป็นรูปสามเหลี่ยม
อาการ ภาวะศีรษะสามหัว
อาการหลักของภาวะศีรษะสามหัวคือการเปลี่ยนแปลงรูปร่างและลักษณะของศีรษะ:
- เมื่อมองจากเหนือยอดศีรษะ กะโหลกศีรษะจะมีรูปร่างคล้ายสามเหลี่ยม
- หน้าผากแคบ;
- สันที่เห็นได้ชัดหรือสัมผัสได้ (กระดูกยื่นออกมา) ซึ่งพาดอยู่ตรงกลางหน้าผาก ทำให้กระดูกหน้าผากมีลักษณะแหลม (คล้ายกระดูกงู)
- ความผิดปกติของส่วนบนของเบ้าตา (สันเหนือเบ้าตาแบนลง) และระยะห่างระหว่างดวงตาต่ำลง (ระยะห่างระหว่างดวงตาลดลง)
กระหม่อมส่วนหน้าอาจปิดก่อนเวลาอันควรได้เช่นกัน
ในภาวะศีรษะสามหัวแบบซินโดรมมีความผิดปกติและสัญญาณของความบกพร่องทางจิตอื่นๆ ในเด็ก [ 16 ]
ภาวะแทรกซ้อนและผลกระทบ
การวินิจฉัย ภาวะศีรษะสามหัว
ภาวะศีรษะสามหัวได้รับการวินิจฉัยตั้งแต่แรกเกิดหรือภายในไม่กี่เดือนหลังคลอด อย่างไรก็ตาม อาจไม่ตรวจพบอาการรุนแรงของภาวะกะโหลกศีรษะปิดไม่สนิทจนกว่าจะถึงช่วงวัยเด็กตอนต้น
เพื่อแสดงภาพพยาธิสภาพของกะโหลกศีรษะ การวินิจฉัยด้วยเครื่องมือจะดำเนินการโดยใช้เอกซเรย์คอมพิวเตอร์ของศีรษะและอัลตราซาวนด์ [ 19 ], [ 20 ]
การวินิจฉัยที่แตกต่างกัน
การวินิจฉัยแยกโรคมีความจำเป็นเพื่อแยกแยะความผิดปกติแบบกลุ่มอาการจากภาวะกระดูกยึดติดกันแบบแยกส่วน ซึ่งเด็กจะได้รับการทดสอบจีโนไทป์
การรักษา ภาวะศีรษะสามหัว
ในเด็กบางราย อาการเมโทปิกซินอสโทซิสค่อนข้างไม่รุนแรง (เมื่อมีร่องที่หน้าผากเพียงเล็กน้อยและไม่มีอาการอื่นใด) ซึ่งไม่จำเป็นต้องได้รับการรักษาเฉพาะ [ 21 ]
การรักษาอาการศีรษะเอียงข้างซ้ายอย่างรุนแรงต้องทำโดยการผ่าตัดเพื่อแก้ไขรูปร่างศีรษะและให้สมองเจริญเติบโตตามปกติ รวมไปถึงการผ่าตัดเพื่อแก้ไขความผิดปกติของกระดูกใบหน้า [ 22 ]
ขั้นตอนการผ่าตัดนี้ ได้แก่ การตัดเนื้อเยื่อเย็บปิดรอบกระดูกเบ้าตา การเปลี่ยนขอบเบ้าตา และการผ่าตัดตกแต่งกระโหลกศีรษะ ซึ่งจะดำเนินการก่อนอายุ 6 เดือน โดยเด็กจะได้รับการตรวจติดตามจนถึงอายุ 1 ขวบ ในช่วงปีแรกของชีวิต เด็กจะได้รับการตรวจเป็นระยะเพื่อให้แน่ใจว่าไม่มีปัญหาเกี่ยวกับการพูด ทักษะการเคลื่อนไหว หรือพฤติกรรม [ 23 ]
การป้องกัน
ไม่มีวิธีการป้องกันความผิดปกติแต่กำเนิดนี้ได้ แต่การปรึกษาทางพันธุกรรมสามารถป้องกันการเกิดเด็กที่มีโรคทางสมองและกะโหลกศีรษะที่รักษาไม่หายได้
ภาวะกะโหลกศีรษะปิดไม่สนิทในทารกในครรภ์สามารถตรวจพบได้โดยการทำอัลตราซาวด์ศีรษะก่อนคลอดในไตรมาสที่ 2 และ 3 ของการตั้งครรภ์
พยากรณ์
การพยากรณ์โรคส่วนใหญ่ขึ้นอยู่กับระดับความผิดปกติของกะโหลกศีรษะ ซึ่งส่งผลต่อการทำงานของระบบประสาทและการรับรู้ของสมอง และหากไม่ได้ทำการผ่าตัดแก้ไข เด็กที่เป็นโรคศีรษะสามหัวจะมีความสามารถทางการรับรู้ทั่วไปต่ำกว่าเด็กวัยเดียวกันที่มีสุขภาพแข็งแรง รวมถึงมีปัญหาในการพูด การมองเห็น ความสนใจ และพฤติกรรม
Использованная литература