Trigonocephaly
ตรวจสอบล่าสุด: 07.08.2022

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
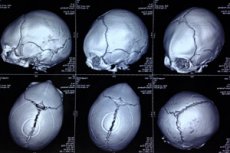
ความผิดปกติ แต่กำเนิดในรูปแบบของกะโหลกศีรษะที่ผิดรูปซึ่งหัวของทารกมีรูปร่างผิดปกติและกะโหลกศีรษะมีลักษณะเป็นรูปสามเหลี่ยมถูกกำหนดให้เป็น trigonocephaly (จากกรีก trigonon - สามเหลี่ยมและ kephale - หัว) [1]
ระบาดวิทยา
ความชุกของ craniosynostosis ประมาณห้ากรณีต่อ 10,000 การเกิดมีชีพ (หรือหนึ่งกรณีต่อ 2-2.5 พันคนในประชากรทั่วไป) [2]
ใน 85% ของกรณี craniosynostosis เป็นระยะ ๆ กรณีที่เหลือเกิดขึ้นเป็นส่วนหนึ่งของกลุ่มอาการ [3]
ตามสถิติการหลอมรวมก่อนวัยอันควรของรอยประสานหน้าผากมัธยฐานเป็นรูปแบบที่พบบ่อยที่สุดอันดับสองของ craniosynostosis และ trigonocephaly คิดเป็นหนึ่งกรณีต่อ 5-15,000 ทารกแรกเกิด; จำนวนทารกเพศชายที่มีอาการผิดปกตินี้สูงกว่าเด็กแรกเกิดเกือบสามเท่า [4]
ในประมาณ 5% ของกรณีนี้ ความผิดปกติแต่กำเนิดนี้มีอยู่ในประวัติครอบครัว [5]
สาเหตุ ไตรโกโนเซฟาลี
การก่อตัวตามปกติของกะโหลกศีรษะเกิดขึ้นเนื่องจากการมีอยู่ของศูนย์การเจริญเติบโตขั้นต้นและการเปลี่ยนแปลงของกระดูก - synarthroses craniofacial (ข้อต่อ) ซึ่งในกระบวนการ ของการพัฒนาโครงกระดูกของศีรษะ ปิดในช่วงเวลาหนึ่งทำให้กระดูกหลอมรวม [6]
กระดูกหน้าผาก (os frontale) ของกะโหลกศีรษะของทารกแรกเกิด ประกอบด้วยสองส่วนระหว่างที่มีการเชื่อมต่อเส้นใยแนวตั้ง - มัธยฐานหน้าผากหรือรอยประสาน metopic (จาก metopon กรีก - หน้าผาก) วิ่งจากด้านบนของด้านหลัง จมูกขึ้นไปกึ่งกลางหน้าผากถึงกระหม่อมหน้า นี่เป็นการเย็บกะโหลกที่มีเส้นใยเพียงเส้นเดียวที่รักษาได้ในช่วงวัยทารก: จาก 3-4 เดือน ถึง 8-18 [7]
ดูเพิ่มเติม - การเปลี่ยนแปลงในกะโหลกศีรษะหลังคลอด
สาเหตุของ trigonocephaly คือ metopic craniosynostosis (craniostenosis) หรือ metopic synostosis (จากภาษากรีก syn - ร่วมกันและ osteon - กระดูก) นั่นคือก่อนวัยอันควร (จนถึงเดือนที่สาม) การรวมตัวของกระดูกของหลุมฝังศพกะโหลกเข้าด้วยกันไม่ได้ รอยประสานหน้าผากมัธยฐาน ดังนั้น craniosynostosis และ trigonocephaly จึงมีความเกี่ยวข้องกันในฐานะเหตุและผล หรือเป็นกระบวนการทางพยาธิวิทยาและผลลัพธ์ของมัน [8]
ในกรณีส่วนใหญ่ trigonocephaly ในเด็กเป็นผลมาจาก craniosynostosis หลัก (ที่แยกได้) ซึ่งไม่ทราบสาเหตุที่แน่ชัด craniosynostosis ที่แยกออกมาเป็นระยะ ๆ ซึ่งอาจเกิดจากการสัมผัสกับปัจจัยทางพันธุกรรมและสิ่งแวดล้อม [9]
แต่ trigonocephaly อาจเป็นส่วนหนึ่งของกลุ่มอาการที่มีมาแต่กำเนิดซึ่งเป็นผลมาจากความผิดปกติของโครโมโซมและการกลายพันธุ์ในยีนต่างๆ ซึ่งรวมถึง: กลุ่มอาการ Opitz trigonocephaly (กลุ่มอาการ Bohring-Opitz), กลุ่มอาการ Apert , กลุ่มอาการ Lois-Dietz, กลุ่มอาการไฟเฟอร์, กลุ่มอาการ Jackson-Weiss, dysostosis กะโหลกศีรษะหรือ Crouzon syndrome , Jacobsen, Setre-Chotzen, Müncke syndromes ในกรณีเช่นนี้ trigonocephaly จะเรียกว่าซินโดรม [10]
เมื่อแรกเกิด สมองมักจะมีขนาด 25% ของขนาดผู้ใหญ่ เข้าถึงสมองของผู้ใหญ่ประมาณ 75% ภายในสิ้นปีแรกของชีวิต แต่ด้วยการชะลอการเจริญเติบโตเบื้องต้นของสมอง ที่เรียกว่า craniosynostosis ทุติยภูมิจึงเป็นไปได้ สาเหตุของความล่าช้านั้นสัมพันธ์กับความผิดปกติของการเผาผลาญ, โรคทางโลหิตวิทยา, ผลการก่อมะเร็งในครรภ์ของสารเคมี (รวมถึงสิ่งที่มีอยู่ในยา) [11]
ผู้เชี่ยวชาญกล่าวว่า trigonocephaly ในผู้ใหญ่ที่ไม่ได้รับการรักษาในวัยเด็กอันเป็นผลมาจาก craniosynostosis ที่แยกได้หรือกลุ่มอาการที่มีมา แต่กำเนิดยังคงมีอยู่ตลอดชีวิต [12]
ปัจจัยเสี่ยง
ผู้เชี่ยวชาญพิจารณาว่าปัจจัยทางพันธุกรรมเป็นปัจจัยเสี่ยงหลักสำหรับ trigonocephaly (และ metopic craniosynostosis เป็นสาเหตุของโรค): ในช่วงสองทศวรรษที่ผ่านมา มีการระบุยีนมากกว่า 60 ยีนที่มีการกลายพันธุ์ที่เกี่ยวข้องกับการหลอมรวมของกระดูกกะโหลกที่ไม่สามารถเคลื่อนที่ได้ก่อนวัยอันควรในทารก
มีความเสี่ยงเพิ่มขึ้นในการเกิดพยาธิสภาพของ craniofacial synarthrosis และ osteogenesis ทั่วไป (การสร้างกระดูก) ในกรณีที่ทารกในครรภ์มีภาวะขาดออกซิเจนในมดลูก การตั้งครรภ์หลายครั้ง การดื่มแอลกอฮอล์ ยาเสพติด หรือการสูบบุหรี่ระหว่างคลอดบุตร [13]
กลไกการเกิดโรค
ตามทฤษฎีที่แพร่หลาย พยาธิกำเนิดของ trigonocephaly อยู่ในการละเมิดการสร้างกระดูกของทารกในครรภ์ในการตั้งครรภ์ระยะแรก ส่วนใหญ่มักเกิดจากปัจจัยทางพันธุกรรม เนื่องจากการตรวจพบความผิดปกติของโครโมโซมแบบสุ่มในทารกแรกเกิดที่มี craniosynostosis metopic ตัวอย่างเช่น หนึ่งในอาการที่พบบ่อยที่สุดคือ trisomy 9p ซึ่งนำไปสู่ความบกพร่องของกะโหลกศีรษะและหน้าและโครงกระดูก ความบกพร่องทางสติปัญญา และการพัฒนาของจิต [14]
เนื่องจากการรวมตัวเร็วเกินไปของรอยประสานหน้าผากมัธยฐาน การเจริญเติบโตในบริเวณนี้ของกะโหลกศีรษะเป็นเรื่องยาก: การเจริญเติบโตด้านข้างของกระดูกหน้าผากถูก จำกัด ด้วยการทำให้แอ่งกะโหลกหน้าสั้นลง สันกระดูกถูกสร้างขึ้นตามแนวกึ่งกลางของหน้าผาก มีการบรรจบกันของกระดูกที่ก่อตัวเป็นวงโคจรของดวงตา และความประทับใจของกระดูกชั่วขณะ [15]
แต่การเติบโตของกะโหลกศีรษะในพื้นที่อื่น ๆ ยังคงดำเนินต่อไป: มีการชดเชยทัล (anteroposterior) และการเติบโตตามขวางของด้านหลังของกะโหลกศีรษะ (ด้วยการขยายตัวของส่วนข้างขม่อม - ท้ายทอย) เช่นเดียวกับการเติบโตในแนวตั้งและทัลของใบหน้า. อันเป็นผลมาจากการละเมิดเหล่านี้กะโหลกศีรษะได้รับรูปร่างผิดปกติ - สามเหลี่ยม
อาการ ไตรโกโนเซฟาลี
อาการหลักของ trigonocephaly คือการเปลี่ยนแปลงรูปร่างและลักษณะของศีรษะ:
- เมื่อมองจากด้านบนหัวกะโหลกจะมีรูปทรงสามเหลี่ยม
- หน้าผากแคบ
- สันที่เห็นได้ชัดเจนหรือมองเห็นได้ (ส่วนที่ยื่นออกมาของกระดูก) วิ่งไปตามกึ่งกลางหน้าผาก ทำให้กระดูกหน้าผากมีรูปร่างแหลม (กระดูกงู)
- ความผิดปกติของส่วนบนของวงโคจร (การแบนของสันเขาเหนือออร์บิทัล) และภาวะ hypotelorism (ลดระยะห่างระหว่างดวงตา)
กระหม่อมด้านหน้า (ด้านหน้า) อาจถูกปิดก่อนเวลาอันควร
เมื่อมีอาการ trigonocephaly เป็นกลุ่มอาการ มีความผิดปกติและสัญญาณ ของภาวะปัญญาอ่อนในเด็กอื่นๆ [16]
การวินิจฉัย ไตรโกโนเซฟาลี
Trigonocephaly ได้รับการวินิจฉัยตั้งแต่แรกเกิดหรือภายในไม่กี่เดือนหลังจากที่ทารกเกิด อย่างไรก็ตาม การค้นพบที่เด่นชัดน้อยกว่าของ metopic craniosynostosis อาจยังคงตรวจไม่พบจนถึงวัยเด็ก
เพื่อให้เห็นภาพพยาธิสภาพของกะโหลกศีรษะการวินิจฉัยด้วยเครื่องมือจะดำเนินการโดยใช้การตรวจเอกซเรย์คอมพิวเตอร์ของศีรษะอัลตราซาวนด์ [19], [20]
การวินิจฉัยที่แตกต่างกัน
การวินิจฉัยแยกโรคเป็นสิ่งที่จำเป็นในการแยกแยะความบกพร่องทางกลุ่มอาการจากซินโทโทซิสเมโทปิกที่แยกได้ ซึ่งเด็กจะได้รับ การวิเคราะห์จีโนไทป์
การรักษา ไตรโกโนเซฟาลี
ในเด็กบางคน กรณีของ metopic synostosis นั้นไม่รุนแรงพอ (เมื่อมีเพียงร่องที่สังเกตได้บนหน้าผากและไม่มีอาการอื่นใด) ที่ไม่ต้องการการรักษาเฉพาะ [21]
การรักษาภาวะศีรษะศรีษะรุนแรงเป็นการผ่าตัด และประกอบด้วยการผ่าตัดเพื่อแก้ไขรูปร่างของศีรษะและดูแลการเจริญเติบโตของสมองตามปกติ ตลอดจนการแก้ไขความผิดปกติของกระดูกใบหน้า [22]
การผ่าตัดดังกล่าว - synostectomy ของ metopic suture, displacement ของ orbital margin และ cranioplasty - ดำเนินการเมื่ออายุไม่เกิน 6 เดือน เด็กอยู่ภายใต้การดูแลจนถึงอายุหนึ่งปี ในช่วงปีแรกของชีวิตเด็ก พวกเขาจะได้รับการตรวจสอบเป็นระยะเพื่อให้แน่ใจว่าไม่มีปัญหาเกี่ยวกับคำพูด ทักษะการเคลื่อนไหว หรือพฤติกรรม [23]
การป้องกัน
วิธีการป้องกันข้อบกพร่องที่เกิดนี้ยังไม่ได้รับการพัฒนา แต่การให้คำปรึกษาทางพันธุกรรมสามารถป้องกันการคลอดบุตรที่มีพยาธิสภาพของกะโหลกศีรษะที่รักษาไม่หาย
และเป็นไปได้ที่จะระบุ craniosynostosis ในทารกในครรภ์ระหว่างอัลตราซาวนด์ก่อนคลอดของศีรษะในไตรมาสที่สองและสามของการตั้งครรภ์
พยากรณ์
การพยากรณ์โรคขึ้นอยู่กับระดับของความผิดปกติของกะโหลกศีรษะในหลาย ๆ ด้าน ซึ่งส่งผลต่อการทำงานของระบบประสาทของสมอง และหากไม่ได้รับการผ่าตัดแก้ไข เด็กที่มีภาวะสมองเสื่อม (trigonocephaly) เมื่อเทียบกับเพื่อนที่มีสุขภาพดี จะมีความรู้ความเข้าใจทั่วไปต่ำกว่า มีปัญหาด้านการพูด การมองเห็น ความสนใจ และพฤติกรรม
Использованная литература