ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
อัลแคปโตนูเรีย คือความผิดปกติของเอนไซม์แต่กำเนิด
ตรวจสอบล่าสุด: 04.07.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
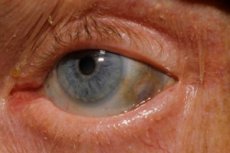
โรคอัลแคปโตนูเรีย ซึ่งเป็นความผิดปกติแต่กำเนิดของการเผาผลาญกรดอะมิโนไทโรซีน เป็นหนึ่งในโรคทางระบบเผาผลาญที่พบได้น้อยมาก
โรคนี้อาจเรียกอีกอย่างหนึ่งว่าภาวะขาดเอนไซม์โฮโมเจนติเซทออกซิเดส โฮโมเจนติซินูเรีย โรคตาบอดสีทางพันธุกรรม หรือโรคปัสสาวะสีดำ[ 1 ]
ระบาดวิทยา
ตามสถิติพบว่ามีผู้ป่วยอัลคาปโตนูเรียไม่เกิน 9 รายต่อประชากร 1 ล้านคน และในประเทศยุโรปส่วนใหญ่พบผู้ป่วย 1 รายต่อทารกเกิดมีชีวิต 100,000-250,000 ราย
ในประเทศยุโรป สโลวาเกีย (โดยเฉพาะภูมิภาคตะวันตกเฉียงเหนือที่มีขนาดค่อนข้างเล็ก) ถือเป็นข้อยกเว้น โดยอัตราการเกิดอัลคาปโตนูเรียอยู่ที่ 1 รายต่อทารกแรกเกิด 19,000 คน ซึ่งสาเหตุส่วนใหญ่น่าจะมาจากครอบครัวชาวโรมานีในสโลวาเกียที่อาศัยอยู่ที่นั่น อัตราการผสมพันธุ์กันในสายเลือดเดียวกัน (การแต่งงานระหว่างลูกพี่ลูกน้อง) สูงที่สุดในยุโรป โดยอยู่ที่ 10-14% [ 2 ]
สาเหตุ โรคอัลคาปโตนูเรีย
สาเหตุที่แน่ชัดของอัลคาปโตนูเรีย ซึ่งเป็นความผิดปกติแต่กำเนิดของการสลายของอะโรมาติก (โฮโมไซคลิก) แอลฟา-อะมิโนแอซิดไทโรซีน ได้รับการยืนยันแล้ว: ความผิดปกติทางเมตาบอลิซึมประเภทนี้เป็นผลมาจากการกลายพันธุ์แบบโฮโมไซกัสหรือแบบผสมเฮเทอโรไซกัสของยีนหนึ่งในหลายพันยีนบนโครโมโซม 3 หรือพูดให้ชัดเจนกว่านั้นคือยีน HGD ที่ตำแหน่ง 3q21-q23 บนแขนยาวของโครโมโซม ยีนนี้เข้ารหัสลำดับนิวคลีโอไทด์ของเอนไซม์ในตับโฮโมเจนติเซท-1,2-ไดออกซิเจเนส [ 3 ] (เรียกอีกอย่างว่าโฮโมเจนติซิกแอซิดออกซิเดสหรือโฮโมเจนติเซทออกซิเดส) ซึ่งเป็นเมทัลโลโปรตีนที่มีธาตุเหล็กซึ่งจำเป็นสำหรับขั้นตอนหนึ่งของการสลายไทโรซีนในร่างกาย [ 4 ], [ 5 ]
ดังนั้นภาวะอัลแคปโทนูเรียคือความบกพร่องของเอนไซม์โฮโมเจนติเซท-1,2-ไดออกซิเจเนส หรือพูดให้ชัดเจนกว่านั้น ก็คือ เป็นผลจากการขาดหรือไม่มีเลยตามที่กำหนดทางพันธุกรรม [ 6 ]
โรคอัลแคปโตนูเรียเป็นภาวะที่ร่างกายขาดเอนไซม์แต่กำเนิด จึงถ่ายทอดลักษณะทางพันธุกรรมที่ถ่ายทอดทางยีนลักษณะด้อย นั่นคือ หากเด็กจะเกิดโรคอัลแคปโตนูเรียได้ พ่อแม่ทั้งสองฝ่ายจะต้องมียีนที่ได้รับการดัดแปลงสำหรับเอนไซม์ เนื่องจากพ่อแม่แต่ละคนจะถ่ายทอดยีนเพียงสำเนาเดียวจากทั้งหมด 2 สำเนาให้กับลูก
ตามข้อมูลล่าสุด มีการดัดแปลงยีน HGD มากกว่าสองร้อยรูปแบบ โดยการกลายพันธุ์แบบมิสเซนส์ การเคลื่อนย้าย และการตัดต่อเป็นสิ่งที่พบเห็นบ่อยที่สุด
ปัจจัยเสี่ยง
ปัจจัยเสี่ยงเพียงประการเดียวสำหรับการพัฒนาโรคเอนไซม์ผิดปกติแต่กำเนิดนี้คือการปรากฏตัวของโรคนี้ในประวัติครอบครัวและการถ่ายทอดยีน HGD ที่ได้รับการดัดแปลงสองชุด หากพ่อแม่ไม่มีภาวะอัลแคปโตนูเรีย (ความเสี่ยงในการถ่ายทอดความผิดปกติอยู่ที่ 25%) หรือพ่อแม่ฝ่ายหนึ่งมีความผิดปกตินี้ [ 7 ]
กลไกการเกิดโรค
ไทโรซีนมีบทบาทสำคัญในการสังเคราะห์โปรตีน การผลิตโครโมโปรตีน ซึ่งเป็นเม็ดสีเมลานินของผิวหนัง รวมถึงฮอร์โมนไทรอยด์และสารสื่อประสาทคาเทโคลามีน
กลไกในการควบคุมปริมาณไทโรซีนในเซลล์มีความซับซ้อนมาก และร่างกายจะทำให้ปริมาณไทโรซีนส่วนเกินเป็นปกติโดยการย่อยสลาย กระบวนการย่อยสลายไทโรซีน เช่นเดียวกับกรดอะมิโนอะโรมาติกทั้งหมด มีหลายขั้นตอนและเกิดขึ้นหลายขั้นตอน แต่ละขั้นตอนของการย่อยสลายไทโรซีนจะเกิดขึ้นโดยมีเอนไซม์เฉพาะเข้ามาเกี่ยวข้องและเกิดสารประกอบตัวกลางขึ้น
ดังนั้น กรดอะมิโนจะถูกย่อยสลายเป็นพารา-ไฮดรอกซีฟีนิลไพรูเวตก่อน จากนั้นจึงเปลี่ยนเป็นกรดอัลคาปโทน – 2,5-ไดไฮดรอกซีฟีนิลอะซิติกหรือกรดโฮโมเจนติซิก จากนั้นอัลคาปโทนจะถูกเปลี่ยนเป็นกรดมาเลอะซิติก แต่สิ่งนี้จะไม่เกิดขึ้น [ 8 ]
การเกิดโรคอัลแคปโทนูเรียประกอบด้วยการหยุดปฏิกิริยาทางชีวเคมีของการสลายตัวของไทโรซีนในระยะการสร้างกรดโฮโมเจนติซิก ซึ่งไม่มีความจำเป็นในการใช้เอนไซม์ที่เรียกว่าโฮโมเจนติเซตออกซิเดสในการย่อยสลายกรดนี้
ร่างกายไม่สามารถใช้กรดโฮโมเจนติซิกได้และสามารถสะสมได้ด้วยการขับถ่ายทางไต นอกจากนี้ กรดยังถูกออกซิไดซ์เป็นเบนโซควิโนอะซิเตท (กรดเบนโซควิโนนอะซิติก) ซึ่งเมื่อจับกับโมเลกุลของเนื้อเยื่อและของเหลวในร่างกาย จะเกิดสารประกอบไบโอโพลีเมอร์ที่มีสีคล้ายเมลานิน
การสะสมของผลิตภัณฑ์กลางเหล่านี้ในเนื้อเยื่อทำให้เกิดการหยุดชะงักของโครงสร้างคอลลาเจนของเนื้อเยื่อกระดูกอ่อน ส่งผลให้ความยืดหยุ่นลดลง ส่งผลให้เกิดอาการทางคลินิกหลายอย่างของภาวะอัลแคปโตนูเรีย และอาจเกิดภาวะแทรกซ้อนได้
อาการ โรคอัลคาปโตนูเรีย
ภาวะอัลแคปโตนูเรียในทารกแรกเกิดและทารกมีลักษณะเฉพาะคือปัสสาวะมีสีเข้มขึ้น เมื่อปัสสาวะสัมผัสกับอากาศ ปัสสาวะบนผ้าอ้อม ผ้าอ้อมเด็ก และชุดชั้นในจะเปลี่ยนเป็นสีน้ำตาลเข้ม เนื่องมาจากกรดโฮโมเจนติซิกสะสมและถูกปล่อยออกมา ซึ่งจะถูกออกซิไดซ์เป็นเบนโซควิโนอะซิเตท [ 9 ]
หากไม่มีอาการอื่น ๆ ภาวะอัลคาปโตนูเรียในเด็กเล็กมักไม่ถูกตรวจพบในเวลาที่เหมาะสม เนื่องจากปัสสาวะอาจเปลี่ยนเป็นสีคล้ำหลังจากปัสสาวะเป็นเวลาหลายชั่วโมง ตามข้อมูลบางส่วน พบว่าเด็กอายุต่ำกว่า 12 เดือนที่เกิดมาพร้อมกับภาวะขาดเอนไซม์นี้เพียงหนึ่งในห้าเท่านั้นที่ได้รับการตรวจพบในสถานพยาบาล ดังนั้น จึงเป็นสิ่งสำคัญมากที่ผู้ปกครองจะต้องใส่ใจในการดูแลทารกของตน
นอกจากนี้ สัญญาณเริ่มต้นยังได้แก่ การมีเม็ดสี (สีเทาอมฟ้า) ของส่วนแข็งของตาและกระดูกอ่อนของหูและจมูก ซึ่งมักเรียกว่าภาวะออโครโนซิส[ 10 ]
เมื่อเวลาผ่านไป อาการอื่น ๆ จะปรากฏขึ้น:
- สีผิวมีสีเข้มมากบริเวณโหนกแก้ม รักแร้ และอวัยวะเพศ
- การเปื้อนเสื้อผ้าเมื่อสัมผัสกับบริเวณที่มีเหงื่อไหลตามร่างกาย
- การโจมตีของความอ่อนแอทั่วไป
- เสียงแหบแห้ง
ควรทราบว่าภาวะอัลแคปโตนูเรียและภาวะโอโครโนซิสดังที่กล่าวข้างต้น เป็นชื่อพ้องสำหรับความผิดปกติของการย่อยสลายไทโรซีนชนิดเดียวกัน
โรคปัสสาวะผสมน้ำเชื่อมเมเปิ้ลและภาวะอัลคาโปนิอูเรีย โรคปัสสาวะผสมน้ำเชื่อมเมเปิ้ลแต่กำเนิดหรือภาวะลิวซิโนซิสเป็นความผิดปกติของระบบเผาผลาญเช่นกัน มีรูปแบบการถ่ายทอดทางพันธุกรรมเหมือนกัน และอาจมีการกลายพันธุ์บนโครโมโซมเดียวกัน แต่ส่งผลต่อยีนที่เข้ารหัสเอนไซม์คอมเพล็กซ์ของเอนไซม์อัลฟาคีโตดีไฮโดรจีเนสโซ่กิ่ง ดังนั้น ร่างกายจึงไม่สามารถย่อยส่วนประกอบบางส่วนของโปรตีนได้ โดยเฉพาะกรดอะมิโนลิวซีน ไอโซลิวซีน และวาลีน เมื่อเป็นโรคนี้ ปัสสาวะ (และขี้หู) จะมีกลิ่นหอม นอกจากนี้ ภาพทางคลินิกของภาวะกรดอินทรีย์ในเลือดชนิดนี้ยังรวมถึงการสร้างเม็ดสีลดลง ความดันโลหิตผันผวน ชัก อาเจียนและท้องเสีย ระดับน้ำตาลในเลือดลดลง กรดคีโตนในเลือด ประสาทหลอน เป็นต้น อัตราการเสียชีวิตในเด็กค่อนข้างสูง ในผู้ใหญ่ หากไม่ได้รับการรักษา อาจเกิดอาการโคม่าและเสียชีวิตเนื่องจากสมองบวมได้
โรคเผือกและอัลแคปโตนูเรียมีไทโรซีนเป็นองค์ประกอบหลักเท่านั้นโรคเผือกรวมถึงโรคผิวหนังรอบดวงตาเกิดจากการกลายพันธุ์ทางพันธุกรรมที่ส่งผลต่อการสร้างเม็ดสีเมลานิน การเปลี่ยนแปลงแต่กำเนิดพบได้ในยีน TYR บนโครโมโซม 11 (11q14.3) ซึ่งเข้ารหัสไทโรซิเนส ซึ่งเป็นเอนไซม์เมลานินที่มีทองแดงเป็นองค์ประกอบที่จำเป็นต่อการสร้างเม็ดสีผิวจากผลผลิตของไทโรซีนที่เผาผลาญ โรคนี้พบได้บ่อยกว่าโรคอัลแคปโตนูเรียมาก
ภาวะแทรกซ้อนและผลกระทบ
ภาวะอัลแคปโทนูเรียเกิดขึ้นจากการทำงานของเมตาบอไลต์ตัวกลางของไทโรซีน - กรดโฮโมเจนติซิกและกรดเบนโซควิโนนอะซิติก - ผลที่ตามมาและภาวะแทรกซ้อนปรากฏเนื่องมาจากการสะสมของโพลีเมอร์ที่มีเม็ดสีที่มีปฏิกิริยา การทำลายของเส้นใยคอลลาเจน และการเสื่อมลงของสภาพของกระดูกอ่อน (โดยที่ความต้านทานต่อแรงกดดันทางกลลดลง)
เมื่อเข้าสู่วัยผู้ใหญ่ โรคข้อเสื่อมและข้อเข่าเสื่อมของข้อใหญ่ (สะโพก กระดูกเชิงกราน และเข่า) จะค่อยๆ พัฒนาขึ้น ช่องว่างระหว่างกระดูกสันหลังจะแคบลง (โดยเฉพาะบริเวณเอวและกระดูกสันหลังส่วนอก) โดยจะมีการสะสมของแคลเซียมและกระดูกงอก ความหนาแน่นของเนื้อเยื่อของแผ่นกระดูกใต้กระดูกอ่อนจะลดลง และกระดูกข้างใต้จะเกิดการเปลี่ยนแปลงทางพยาธิวิทยาพร้อมกับการเจริญเติบโตและการเสียรูป [ 11 ]
อาจพบความเสียหายต่อลิ้นหัวใจ (ลิ้นหัวใจเอออร์ติกและลิ้นหัวใจไมทรัล) และหลอดเลือดหัวใจ – โดยมีสัญญาณของโรคหลอดเลือดหัวใจ รวมถึงการก่อตัวของนิ่วในไตและต่อมลูกหมาก – เนื่องมาจากการสะสมของแคลเซียมชนิดเดียวกัน [ 12 ], [ 13 ]
การวินิจฉัย โรคอัลคาปโตนูเรีย
โดยทั่วไป การวินิจฉัยความผิดปกติทางการเผาผลาญแต่กำเนิดจะขึ้นอยู่กับการศึกษาของเหลวในร่างกาย
การวินิจฉัยภาวะอัลแคปโทนูเรียสามารถวินิจฉัยได้จากการทดสอบและปฏิกิริยาใดบ้าง? จำเป็นต้องตรวจปัสสาวะเพื่อตรวจหากรดโฮโมเจนติซิกและกำหนดระดับของกรดดังกล่าว (ปกติ – 20-30 มก. ต่อวัน สูงขึ้น – 3-8 ก.) ตรวจตัวอย่างปัสสาวะโดยใช้แก๊สโครมาโทกราฟีหรือแมสสเปกโตรเมตรี โดยใช้โครมาโทกราฟีของเหลว การทดสอบคัดกรองเพื่อหาปริมาณเหล็กคลอไรด์ในปัสสาวะก็เป็นไปได้ [ 14 ]
ยังมีวิธีการวินิจฉัยอย่างรวดเร็วอีกด้วย โดยการตรวจสอบอัลคาปตอนในคราบปัสสาวะแห้งบนกระดาษ (โดยดูจากความเข้มข้นของสี)
ในการชี้แจงการวินิจฉัย การวินิจฉัยด้วยเครื่องมือ (รังสีวิทยา) เกี่ยวข้องกับการระบุสัญญาณทางรังสีของโรคข้อเข่าเสื่อมและพยาธิสภาพของข้ออื่น ๆ ในผู้ป่วย
การวินิจฉัยได้รับการยืนยันด้วยวิธีการทางพันธุศาสตร์โมเลกุลในการวินิจฉัยโรคทางพันธุกรรม เช่น การทดสอบทางพันธุกรรมและการจัดลำดับดีเอ็นเอ [ 15 ]
การวินิจฉัยที่แตกต่างกัน
การวินิจฉัยแยกโรค ได้แก่ ภาวะเม็ดเลือดแดงเข้มและภาวะตับวายเฉียบพลันของทารกแรกเกิด ภาวะเมลานินในปัสสาวะ ภาวะพอร์ฟิเรียเฉียบพลันเป็นระยะๆ ภาวะลิมโฟฮิสโตไซโตซิสในเม็ดเลือด พยาธิสภาพของไมโตคอนเดรียขั้นต้น โรคข้ออักเสบรูมาตอยด์ โรคข้ออักเสบติดกระดูกสันหลัง
ใครจะติดต่อได้บ้าง?
การรักษา โรคอัลคาปโตนูเรีย
การรักษาหลักสำหรับภาวะอัลคาปโตนูเรียคือการให้กรดแอสคอร์บิกในปริมาณมาก (อย่างน้อย 1,000 มก. ต่อวัน) ทางปาก ในเด็ก การทำเช่นนี้จะเพิ่มการขับกรดโฮโมเจนติซิกในปัสสาวะ และในผู้ใหญ่ การทำเช่นนี้จะลดปริมาณกรดเบนโซควิโนนอะซิติกซึ่งเป็นอนุพันธ์ของกรดแอสคอร์บิกในปัสสาวะ และทำให้การจับกับเนื้อเยื่อเกี่ยวพันของข้อต่อและคอลลาเจนช้าลง [ 16 ]
คลินิกในยุโรปตะวันตกกำลังทดสอบยา Nitisinone (Orfalin) ซึ่งเป็นยาจากกลุ่มเมตาบอไลต์ที่ยับยั้งขั้นตอนที่สองของกระบวนการย่อยสลายไทโรซีน ซึ่งก็คือการเปลี่ยนพาราไฮดรอกซีฟีนิลไพรูเวตให้เป็นกรดโฮโมเจนทิซิก อย่างไรก็ตาม การใช้ยาตัวนี้จะทำให้ไทโรซีนสะสมและอาจทำให้เกิดผลข้างเคียงร้ายแรง เช่น กระจกตาขุ่นและกลัวแสง เลือดกำเดาไหลและเลือดออกในกระเพาะ ตับวาย การเปลี่ยนแปลงของเลือด เป็นต้น อย่างไรก็ตาม ในสหรัฐอเมริกา Nitisinone ได้รับการอนุมัติจาก FDA ให้ใช้ในการรักษาไทโรซิเนเมียชนิดที่ 1 [17 ], [ 18 ]
ดังนั้น การบำบัดทางกายภาพบำบัดจึงดำเนินการสำหรับปัญหาข้อที่เกิดจากภาวะอัลแคปโตนูเรีย ได้แก่ การบำบัดด้วยการออกกำลังกายเพื่อเพิ่มความแข็งแรงของกล้ามเนื้อและปรับปรุงการเคลื่อนไหวของข้อต่อ การบำบัดด้วยน้ำเกลือ และการบำบัดด้วยแผ่นแปะเพื่อลดอาการปวด
แม้ว่าไทโรซีนจะไม่ได้มาจากอาหารเท่านั้น แต่ยังผลิตได้ในร่างกายด้วย ผู้ป่วยที่เป็นโรคอัลแคปโตนูเรียควรรับประทานอาหารที่มีโปรตีนต่ำ และจำกัดการรับประทานอาหารที่มีไทโรซีนสูง โดยเฉพาะอย่างยิ่งเนื้อวัวและเนื้อหมู ผลิตภัณฑ์จากนม (โดยเฉพาะชีส) ถั่วและเมล็ดพืช
การป้องกัน
การป้องกันการกลายพันธุ์ของยีนเป็นสิ่งที่เป็นไปไม่ได้ แต่เพื่อป้องกันการเกิดของเด็กที่มีความเสี่ยงสูงต่อความผิดปกติแต่กำเนิด จึงมีการให้คำปรึกษาด้านพันธุกรรมทางการแพทย์ ซึ่งจำเป็นก่อนที่จะวางแผนการตั้งครรภ์สำหรับคู่รักที่มีประวัติครอบครัวที่มีโรคทางพันธุกรรม [ 19 ]
พยากรณ์
ผลร้ายแรงจากภาวะอัลแคปโตนูเรียเกิดขึ้นได้ยากมาก และการเสียชีวิตอาจเกิดจากภาวะแทรกซ้อนร้ายแรงที่เกี่ยวข้องกับหัวใจและไต ดังนั้น อายุขัยโดยรวมของผู้ที่เป็นโรคอัลแคปโตนูเรียจึงถือว่าดี
แต่คุณภาพชีวิตจะลดลงเนื่องจากความเจ็บปวดบริเวณข้อหรือกระดูกสันหลังอย่างรุนแรง ส่งผลให้มีการเคลื่อนไหวที่จำกัดและมักจะลุกลามมากขึ้น