ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
โรคฮันติงตัน
ตรวจสอบล่าสุด: 23.04.2024

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
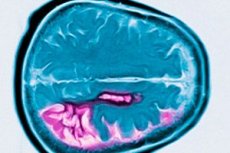
โรคฮันติงตัน- autosomal โรคเกี่ยวกับระบบประสาทที่โดดเด่นเอกลักษณ์เฉพาะด้วยการเริ่มต้นที่อายุเฉลี่ยของความบกพร่องทางสติปัญญาก้าวหน้าเคลื่อนไหวโดยไม่ตั้งใจและพิการประสานงานมอเตอร์ การวินิจฉัยยืนยันโดยการทดสอบทางพันธุกรรม การรักษาอาการส่วนใหญ่ ญาติเลือดสามารถแนะนำให้รับการทดสอบทางพันธุกรรม จอร์จฮันติงตันเป็นคนแรกที่อธิบายสภาพนี้ในปี 1872 หลังจากตรวจสอบกรณีครอบครัวของโรคจากชาวลองไอส์แลนด์
ความชุกของโรคฮันติงตันอยู่ที่ประมาณ 10 รายต่อประชากร 100,000 คนและเมื่อเริ่มมีอาการปลายคนประมาณ 30 คนจาก 100,000 คนมีความเสี่ยง 50% ที่จะได้รับมันในช่วงชีวิตของพวกเขา แม้ว่าโรคส่วนใหญ่มักจะปรากฏตัวเมื่ออายุ 35-40 ปี แต่ช่วงอายุที่เริ่มมีอาการค่อนข้างกว้าง: การโจมตีที่เร็วที่สุดจะถูกบันทึกเมื่ออายุ 3 ปีและล่าสุด - 90 ปี แม้ว่าในตอนแรกเชื่อว่าโรคนี้มีลักษณะทะลุปรุโปร่ง 100% แต่ตอนนี้เชื่อว่าไม่เป็นเช่นนั้น ในบุคคลที่สืบทอดยีนสำหรับโรคจากพ่อโรคจะปรากฏโดยเฉลี่ย 3 ปีก่อนหน้านี้กว่าความสะดวกสบายที่สืบทอดยีนทางพยาธิวิทยาจากแม่ ในเวลาเดียวกันในประมาณ 80% ของผู้ป่วยที่สืบทอดยีนทางพยาธิวิทยาจากพ่อโรคนี้ปรากฏตัวถึง 20 ปี ปรากฏการณ์ของการปรากฏตัวก่อนหน้านี้ของข้อบกพร่องทางพันธุกรรมในลูกหลานเรียกว่าการรอคอย
 [1],
[1],
สาเหตุของโรคฮันติงตันคืออะไร
โรคฮันติงตันไม่มีลักษณะที่อ่อนโยน ฝ่อของนิวเคลียส caudate จะปรากฏขึ้นที่เซลล์ประสาทขนาดเล็กเสื่อมโทรมและระดับของสารสื่อประสาท - gamma-aminobutyric acid (GABA) และสารพีลดลง
ยีนกลายพันธุ์ที่มีจำนวนเพิ่มขึ้น (“ การขยายตัว”) ของลำดับดีเอ็นเอ CAG (cysteine - alanine - glycine), การเข้ารหัสกรดอะมิโนกลูตามีนรับผิดชอบการพัฒนาของโรคฮันติงตัน ผลิตภัณฑ์ของยีนนี้ - gatinging โปรตีนขนาดใหญ่ - มีจำนวนที่เหลือของ polyglutamine ตกค้างซึ่งนำไปสู่โรคโดยกลไกที่ไม่รู้จัก ยิ่งมี CAG ซ้ำมากขึ้นเท่าไหร่โรคก็จะเปิดตัวเร็วขึ้นและหนักขึ้นเท่านั้น จากรุ่นสู่รุ่นจำนวนซ้ำอาจเพิ่มขึ้นซึ่งเมื่อเวลาผ่านไปนำไปสู่การทำให้รุนแรงของฟีโนไทป์ของครอบครัว
แม้จะมีความสนใจอย่างมากในการเปลี่ยนแปลงทางพันธุกรรมและชีวเคมีในโรคพาร์กินสันการค้นหายีนของโรคก็ไม่ประสบความสำเร็จจนถึงปลายปี 1970 ในเวลานี้แนนซี่เว็กซ์เลอร์และอัลลันโทบิน (A. Tobin) จัดประชุมเชิงปฏิบัติการที่ได้รับการสนับสนุนจากมูลนิธิโรคทางพันธุกรรมเพื่อหารือเกี่ยวกับกลยุทธ์ในการค้นหายีนโรคฮันติงตัน David Houseman (D. Housman), David Botstein (D. Votstein) และ Ray White (R. White) ผู้เข้าร่วมประชุมแนะนำว่าเทคนิคการรวมตัวกันของ DNA ที่พัฒนาขึ้นใหม่สามารถช่วยให้บรรลุเป้าหมายนี้ได้ ภารกิจสำคัญในโครงการภายใต้การพัฒนาคือการค้นหาครอบครัวใหญ่ที่สมาชิกต้องทนทุกข์ทรมานจากโรคฮันติงตันในหลายชั่วอายุคนเพื่อรับตัวอย่างดีเอ็นเอ ในปี 1979 โครงการร่วมของนักวิทยาศาสตร์จากเวเนซุเอลาและสหรัฐอเมริกาได้เปิดตัวซึ่งรวมถึงการสำรวจของครอบครัวขนาดใหญ่ที่มีโรคฮันติงตันอาศัยอยู่บนชายฝั่งของทะเลสาบมาราเชโบ (เวเนซุเอลา) ในปี 1983 ยีนของโรคฮันติงตันตั้งอยู่ที่ปลายแขนสั้นของโครโมโซมที่ 4 (Gusella et al., 1983) และหนึ่งทศวรรษต่อมาก็มีการเปิดเผยว่าการกลายพันธุ์ของยีนนี้เป็นการเพิ่มจำนวนซ้ำของไซโตไคน์ - อะดีนีน กลุ่มวิจัยความร่วมมือโรค, 2536) วิธีการที่พัฒนาโดยกลุ่มวิทยาศาสตร์นี้ในปัจจุบันถือว่าเป็นมาตรฐานสำหรับการโคลนนิ่งตำแหน่งของยีนใหม่
ในขณะที่ยีนประเภทป่ามีการยืดซ้ำของ 10-28 CAG, รูปแบบที่กลายพันธุ์ของยีนที่ทำให้เกิดโรคฮันติงตันมีการยืดเพิ่มขึ้นจาก 39 เป็นมากกว่า 100 CAG ซ้ำ การระบุการขยายตัวของ trinucleotide ซ้ำช่วยให้เราสามารถอธิบายลักษณะทางคลินิกของโรค โดยเฉพาะอย่างยิ่งพบความสัมพันธ์แบบผกผันระหว่างอายุที่เริ่มมีอาการและความยาวของไซต์ที่มี trinucleotides ซ้ำ ความคาดหวังของการถ่ายทอดทางพันธุกรรมของพ่อสามารถอธิบายได้ด้วยความจริงที่ว่าการเพิ่มจำนวนซ้ำ ๆ มักเกิดขึ้นในผู้ชายระหว่างการสร้างสเปิร์ม การวิเคราะห์การกลายพันธุ์ใหม่แสดงให้เห็นว่าพวกเขามักจะเกิดขึ้นเมื่อผู้ปกครองคนใดคนหนึ่งโดยปกติพ่อมีจำนวน CAG ซ้ำสูงกว่า 28; ในกรณีนี้จำนวนการทำซ้ำเพิ่มขึ้นในรุ่นต่อไป เป็นที่ยอมรับแล้วว่าหากจำนวนการทำซ้ำไม่เกิน 28 ก็จะถูกส่งอย่างเสถียรจากรุ่นสู่รุ่น หากจำนวนการทำซ้ำอยู่ระหว่าง 29 ถึง 35 อาการของโรคฮันติงตันจะไม่ปรากฏขึ้น แต่เมื่อย้ายไปยังลูกหลานความยาวของบริเวณนี้อาจเพิ่มขึ้น หากจำนวนการทำซ้ำเป็น 36 ถึง 39 ในบางกรณี (แต่ไม่เสมอไป) โรคอาจปรากฏออกมาทางคลินิก (การแทรกซึมที่ไม่สมบูรณ์) และการถ่ายทอดไปยังลูกหลานการเพิ่มจำนวนของทรินิวคลีโอไทด์ซ้ำอาจเกิดขึ้นได้ หากจำนวนการทำซ้ำเกินกว่า 40 แสดงว่าโรคนั้นเกิดขึ้นในเกือบทุกกรณีและหากมีการถ่ายโอนไปยังลูกหลานการขยายตัวของการทำซ้ำอาจเป็นไปได้ สาเหตุของการเพิ่มจำนวนการทำซ้ำยังไม่ทราบ
พยาธิวิทยาของโรคฮันติงตัน
โรคฮันติงตันเป็นลักษณะของการตายของเซลล์ประสาทส่วนใหญ่ในนิวเคลียสหางและเปลือกบางส่วนยังอยู่ในเยื่อหุ้มสมองและโครงสร้างอื่น ๆ ของสมอง น้ำหนักรวมของสมองในโรคฮันติงตันลดลงไม่เพียงโดยการลดจำนวนของเซลล์ประสาท แต่เนื่องจากการสูญเสียของสารสีขาว ในเยื่อหุ้มสมองสมองเซลล์ในชั้น V และ VI ได้รับผลกระทบมากที่สุด ความรุนแรงของการเปลี่ยนแปลงความเสื่อมแบบไมโครและขนาดมหภาค (ด้วยการแก้ไขอายุเมื่อเสียชีวิต) มีความสัมพันธ์กับจำนวนการทำซ้ำ CAG การวิเคราะห์ทางพยาธิวิทยาอย่างละเอียดของการเปลี่ยนแปลงในหลายร้อยกรณีของโรคฮันติงตันแสดงให้เห็นว่าการเสื่อมสภาพของ striatum เริ่มต้นด้วยส่วน dorsomedial ของนิวเคลียสหางและส่วน dorsolateral ของเปลือกแล้วแพร่กระจายไปในทิศทางที่หน้าท้อง กลุ่มเซลล์ต่าง ๆ ของนิวเคลียส caudate และเปลือกไม่ประสบในระดับเดียวกัน เซลล์ประสาทที่ถูกสอดเข้าไปใน striatum ยังคงไม่บุบสลาย ในรูปแบบของโรคฮันติงตันเด็กและเยาวชนการเปลี่ยนแปลงทางพยาธิวิทยาใน striatum มีความเด่นชัดมากขึ้นและพบบ่อยมากขึ้นที่เกี่ยวข้องกับเยื่อหุ้มสมองสมอง, สมองน้อย, สมองน้อย, ฐานดอก, ซีดบอล
การเปลี่ยนแปลงทางเคมีประสาทในโรคฮันติงตัน
GABA การศึกษาทางประสาทวิทยาของสมองในผู้ป่วยโรคฮันติงตันเปิดเผยว่าการลดลงของความเข้มข้นของ GABA ใน striatum การศึกษาครั้งต่อไปยืนยันว่าจำนวนของเซลล์ประสาท GABAergic ลดลงในโรคฮันติงตันและแสดงให้เห็นว่าความเข้มข้นของ GABA นั้นลดลงไม่เพียง แต่ใน striatum เท่านั้น แต่ยังอยู่ในพื้นที่ฉายภาพของมัน - ส่วนนอกและส่วนในของลูกโลกซีด ในสมองของโรคฮันติงตันการเปลี่ยนแปลงของตัวรับ GABA ก็ถูกเปิดเผยโดยใช้การจับตัวรับและในการผสมพันธุ์ในแหล่งกำเนิดของ mRNA จำนวนของตัวรับ GABA ลดลงในระดับปานกลางในนิวเคลียสหางและเปลือก แต่เพิ่มขึ้นในส่วนไขว้กันของนิโกร substantia เนื่องจากการแพ้ denervation
Acetylcholine Acetylcholine ใช้เป็นสารสื่อประสาทสำหรับเซลล์ประสาทขนาดใหญ่ที่ไม่สามารถมองเห็นได้ใน striatum ในการศึกษาหลังการชันสูตรต้นในผู้ป่วยโรคฮันติงตันพบว่ามีการลดลงของกิจกรรม cholinecetyltransferase (HAT)ใน striatum ซึ่งอาจบ่งบอกถึงการสูญเสียของเซลล์ประสาท cholinergic อย่างไรก็ตามเมื่อเปรียบเทียบกับจำนวนเซลล์ประสาท GABAergic ที่ลดลงอย่างมีนัยสำคัญเซลล์ประสาทที่ได้รับ cholinergic ยังคงไม่เปลี่ยนแปลง ดังนั้นความหนาแน่นของเซลล์ประสาทบวก acetylcholinesterase และกิจกรรมของ HAT ใน striatum จึงค่อนข้างสูงเมื่อเทียบกับการควบคุมที่มีความสมดุลของอายุ
สารอาร์สารสารพีมีอยู่ในเซลล์ประสาทสไตลอยด์ขนาดกลางจำนวนมากของ striatum ซึ่งส่วนใหญ่จะถูกฉายลงบนส่วนด้านในของลูกบอลสีซีดและ substantia nigra และมักจะมี dorforph และ GABA ระดับของสาร P ใน striatum และ reticular part ของ substantia nigra จะลดลงในโรคฮันติงตัน ในระยะสุดท้ายของโรคโดยใช้การศึกษาทางอิมมูโนฮิสโตเคมีพบว่าจำนวนของเซลล์ประสาทที่มีสารลดลงอย่างมีนัยสำคัญในช่วงก่อนหน้านี้เซลล์ประสาทที่มีสาร P และฉายลงบนส่วนด้านในของลูกบอลสีซีด
Opioid เปปไทด์ Enkephalin ถูกค้นพบในเซลล์ประสาท GABAergic ในรูปแบบการฉายภาพในลักษณะทางอ้อมโดยฉายไปที่ส่วนด้านนอกของลูกบอลสีซีดและถือตัวรับ D2 ด้วยตนเอง จากการศึกษาทางอิมมูโนวิทยาทางเคมีพบว่าในระยะเริ่มต้นของโรคฮันติงตันมีการสูญเสียของเซลล์ประสาทที่มี enkephalin ที่ยื่นออกไปด้านนอกของลูกกลมสีซีด เห็นได้ชัดว่าเซลล์เหล่านี้ตายเร็วกว่าเซลล์ที่มีสาร P และฉายลงบนส่วนด้านในของลูกบอลสีซีด
Catecholamines เซลล์ประสาทที่ประกอบด้วยไบโอจีนินเอมีน (โดพามีน, เซโรโทนิน) และฉายลงบน striatum นั้นตั้งอยู่ในส่วนที่มีขนาดกะทัดรัดของ substantia nigra, ฝาหน้าท้องและนิวเคลียสเย็บ ในขณะที่การคาดคะเนของ noradrenergic ใน striatum ของมนุษย์มีน้อยที่สุดระดับของ serotonin และ dopamine (ในรูปของกรัมของเนื้อเยื่อ) ใน striatum จะเพิ่มขึ้นซึ่งบ่งบอกถึงความปลอดภัยของการคาดคะเนอวัยวะของเซลล์เหล่านี้ เซลล์ประสาทโดปามีนิกของ substantia นิโกรยังคงสภาพเหมือนเดิมทั้งในรูปแบบคลาสสิคและเยาวชนของโรคฮันติงตัน
Somatostatin / neuropeptide Y และไนตริกออกไซด์ synthetase การวัดระดับของ somatostatin และ neuropeptide Y ใน striatum ในโรคฮันติงตันเปิดเผยว่าเพิ่มขึ้น 4-5 เท่าเมื่อเทียบกับเนื้อเยื่อปกติ โดยใช้การศึกษาทางอิมมูโนเคมีโตพบว่ามีความปลอดภัยอย่างสมบูรณ์ของเซลล์ประสาท striatum interstitial striatum ที่มี neuropeptide Y, somatostatin และ nitric oxide synthetase ดังนั้นเซลล์ประสาทเหล่านี้จึงทนต่อกระบวนการทางพยาธิวิทยา
กรดอะมิโนที่น่าตื่นเต้น มีคนแนะนำว่าการตายของเซลล์ที่เลือกสรรในโรคฮันติงตันนั้นสัมพันธ์กับผลกระทบของพิษต่อระบบประสาทที่เกิดจากกลูตาเมต กลูตาเมตและระดับกรด quinolinic (neurotoxin ภายนอกซึ่งเป็นผลพลอยได้จากการเผาผลาญอาหารของตัวเอก serotonin และเป็น retsptorov glugamatnyh) ใน striatum ของโรคฮันติงตันที่มีการเปลี่ยนแปลงเล็กน้อย แต่การศึกษาล่าสุดที่ใช้ MR -สเปกโทรสโกเปิดเผยในร่างกายเพิ่มขึ้นในกลูตาเมต ระดับของเอนไซม์ glial ที่รับผิดชอบในการสังเคราะห์กรด quinolinic ใน striatum ในโรคฮันติงตันเพิ่มขึ้นประมาณ 5 เท่าเมื่อเทียบกับบรรทัดฐานในขณะที่กิจกรรมของเอนไซม์ที่ช่วยให้การย่อยสลายของกรด quinolinic เพิ่มขึ้นในโรคฮันติงตันเพียง 20-50% ดังนั้นการสังเคราะห์กรด quinolinic ในโรคฮันติงตันสามารถเพิ่มขึ้นได้
การสืบสวนของตัวรับกรดอะมิโน excitatory (HAC)ในโรคฮันติงตันเปิดเผยจำนวนลดลงอย่างมีนัยสำคัญในจำนวนของ NMDA-, AMPA-, kainate และ metabotropic glugamat ผู้รับใน striatum เช่นเดียวกับ AMPA- และตัวรับ kainate ในเยื่อหุ้มสมอง ในช่วงปลายของโรคฮันติงตัน, ตัวรับ NMDA ขาดไปจริง, ในระยะแรกและระยะแรกมีการลดลงอย่างมีนัยสำคัญในจำนวนของตัวรับเหล่านี้
เลือกความไว ในโรคของฮันติงตันเซลล์เซลล์สืบพันธุ์บางชนิดเลือกตาย เซลล์ประสาทสไตลอยด์ระดับกลางที่ยื่นออกไปส่วนนอกของเซลล์ซีดและบรรจุ GABA และ enkephalin แล้วตายในระยะแรกของโรคเช่นเดียวกับเซลล์ประสาทที่ประกอบด้วย GABA และสาร P และฉายลงบนส่วนที่เป็นตาข่ายของ substantia nigra การสูญเสียของเซลล์ประสาทที่มี GABA และ enkephalin และฉายลงบนส่วนด้านนอกของลูกบอลสีซีดปลดอาวุธโครงสร้างนี้ซึ่งในทางกลับกันนำไปสู่การยับยั้งการใช้งานของนิวเคลียส subthalamic การลดลงของกิจกรรมของนิวเคลียส subtalamic สามารถอธิบายได้อย่างชัดเจนโดยการเคลื่อนไหวของ choreiform ที่เกิดขึ้นในโรคฮันติงตัน เป็นที่ทราบกันมานานแล้วว่ารอยโรคโฟกัสของนิวเคลียสของตาตาลามิสามารถเป็นสาเหตุของอาการชักกระตุก การสูญเสียของเซลล์ประสาทที่มี GABA และสาร P และการฉายลงบนส่วนไขว้กันของ substantia นิโกรอาจเป็นสาเหตุของความผิดปกติของกล้ามเนื้อกลมที่พบในโรคฮันติงตัน เส้นทางนี้โดยปกติจะยับยั้งเซลล์ประสาทของส่วนไขว้กันเหมือนแหของ substantia นิโกรฉายบนยอดเนินของรูปสี่เหลี่ยมขนมเปียกปูนซึ่งหันควบคุม saccades ในรูปแบบของโรคฮันติงตันเด็กและเยาวชนเส้นทางดังกล่าวข้างต้นประสบอย่างรุนแรงมากขึ้นและนอกจากนี้การคาดคะเน striatal ไปยังส่วนด้านในของลูกซีดจะหายไปก่อน
โปรตีน Huntingtin ถูกเข้ารหัสโดยยีนซึ่งการกลายพันธุ์ที่ทำให้เกิดโรคฮันติงตันถูกตรวจพบในโครงสร้างสมองและเนื้อเยื่ออื่น ๆ โดยปกติแล้ว Huntingtin จะพบได้ในโปรโตปลาสซึมของเซลล์ประสาท โปรตีนถูกตรวจพบในเซลล์ประสาทส่วนใหญ่ของสมอง แต่เมื่อข้อมูลล่าสุดแสดงเนื้อหาของมันจะสูงกว่าเมทริกซ์ในเซลล์ประสาท striosome และในเซลล์ประสาทที่ฉายนั้นมีค่าสูงกว่าในเซลล์อุกกาบาต ดังนั้นการเลือกความไวของเซลล์ประสาทมีความสัมพันธ์กับเนื้อหาของการล่าสัตว์ในพวกเขาซึ่งมักจะแสดงในประชากรบางเซลล์ประสาท
เช่นเดียวกับในสมองของผู้ป่วยที่มีโรคฮันติงตันในหนูที่ถ่ายยีนส่วนเอ็นเทอร์มินัลของยีนโรคฮันติงตันที่มีจำนวนการทำซ้ำเพิ่มขึ้น Huntingtin สร้างมวลรวมหนาแน่นในนิวเคลียสของเซลล์ประสาท การรวมตัวของ intranuclear เหล่านี้เกิดขึ้นในโครงร่างของเส้นโครงร่าง ในหนูที่ดัดแปลงพันธุกรรมการรวมตัวเป็นเวลาหลายสัปดาห์ก่อนที่จะเริ่มมีอาการ ข้อมูลเหล่านี้บ่งชี้ว่าการล่าโปรตีนตินซึ่งมีจำนวนกลูตามีนตกค้างเพิ่มขึ้นการรวมตัวกันของการเข้ารหัส trinucletide ซ้ำหรือชิ้นส่วนของมันสะสมอยู่ในนิวเคลียสเป็นผลให้การควบคุมฟังก์ชั่นของเซลล์นั้นอาจประสบ
อาการของโรคฮันติงตัน
อายุที่อาการแรกปรากฏขึ้นในผู้ป่วยที่เป็นโรคฮันติงตันนั้นยากที่จะระบุด้วยความแม่นยำเนื่องจากโรคนั้นค่อยๆปรากฏขึ้น การเปลี่ยนแปลงบุคลิกภาพและพฤติกรรมความผิดปกติเล็ก ๆ น้อย ๆ ในการประสานงานอาจเกิดขึ้นได้หลายปีก่อนที่อาการจะเด่นชัดขึ้น ตามเวลาของการวินิจฉัยเคลื่อนไหว choreic การประสานงานบกพร่องของการเคลื่อนไหวที่บอบบางและการชะลอตัวในการสร้าง saccades โดยพลการจะพบในผู้ป่วยส่วนใหญ่ ในขณะที่โรคดำเนินไปความสามารถในการจัดระเบียบกิจกรรมของมันจะลดลงความจำลดลงการพูดกลายเป็นเรื่องยากความบกพร่องทางกล้ามเนื้อและประสิทธิภาพการทำงานที่ผิดปกติของการเคลื่อนไหวประสานงานเพิ่มขึ้น แม้ว่าในระยะแรกของโรคจะไม่มีการเปลี่ยนแปลงของกล้ามเนื้อและท่าทางเนื่องจากความก้าวหน้าของมันท่าทาง dystonic อาจพัฒนาซึ่งเมื่อเวลาผ่านไปสามารถกลายเป็นอาการที่โดดเด่น ในช่วงปลายคำพูดจะไม่สามารถเข้าใจได้การกลืนกลายเป็นเรื่องยากมากขึ้นการเดินเป็นไปไม่ได้ โรคของฮันติงตันมักจะดำเนินไปภายใน 15-20 ปี ในระยะสุดท้ายผู้ป่วยหมดหนทางและต้องการการดูแลอย่างต่อเนื่อง ผลลัพธ์ที่เป็นอันตรายไม่ได้เชื่อมโยงโดยตรงกับโรคหลัก แต่เกิดจากภาวะแทรกซ้อนเช่นปอดบวม
ภาวะสมองเสื่อมในโรคฮันติงตัน
รหัส ICD-10
R02.2 ภาวะสมองเสื่อมในโรคฮันติงตัน (G10)
ภาวะสมองเสื่อมพัฒนาเป็นหนึ่งในอาการของกระบวนการเสื่อม - ระบบตีบด้วยระบบแผลหลักของระบบ striatal ของสมองและนิวเคลียส subcohecal อื่น ๆ สืบทอดโดย autosomal เด่น
ตามกฎโรคปรากฏในทศวรรษที่สามหรือสี่ของชีวิตกับ hyperkinesis choreiform (โดยเฉพาะอย่างยิ่งในใบหน้า, แขน, ไหล่, การเดิน), การเปลี่ยนแปลงบุคลิกภาพ (ตื่นเต้น, ตีโพยตีพายและ schizoid ประเภทของบุคลิกภาพผิดปกติ), โรคจิต (ภาวะซึมเศร้าเฉพาะที่มีความเศร้าโศก อารมณ์หวาดระแวง)
ความสำคัญเป็นพิเศษสำหรับการวินิจฉัยคือการรวมกันของ choreoform hyperkinesis, ภาวะสมองเสื่อมและภาระทางพันธุกรรม ต่อไปนี้เป็นสิ่งเฉพาะสำหรับภาวะสมองเสื่อมนี้:
- ความก้าวหน้าอย่างช้าๆ (เฉลี่ย 10-15 ปี): การแยกความแตกต่างระหว่างความสามารถแบบถาวรเพื่อหารือเกี่ยวกับตนเองและความไม่ลงรอยกันทางปัญญาที่เห็นได้ชัดในสถานการณ์ที่ต้องทำงานด้านจิตใจอย่างมีประสิทธิผล (การคิดเชิงแนวคิดการเรียนรู้สิ่งใหม่);
- ความผิดปกติอย่างรุนแรงของการทำงานทางจิตซึ่งขึ้นอยู่กับการละเมิดขั้นต้นของความสนใจและความไม่แน่นอนของทัศนคติของผู้ป่วย ("ฉับพลัน" ความคิดโดยการเปรียบเทียบกับ hyperkinesis);
- ความผิดปกติของการละเมิดที่ชัดเจนของฟังก์ชั่นเยื่อหุ้มสมองที่สูงขึ้น;
- ความสัมพันธ์ผกผันระหว่างการเพิ่มขึ้นของภาวะสมองเสื่อมและความรุนแรงของโรคจิต
โดยคำนึงถึงสัดส่วนที่สูงของโรคจิต (อาการหวาดระแวงหวาดระแวงความอิจฉาริษยาการประหัตประหาร) และ dysphoric ในภาพทางคลินิกของโรคการรักษาจะดำเนินการโดยใช้อินซูลินต่าง ๆ ที่ปิดกั้นตัวรับ dopaminergic (phenothiazine และ butyrophenone อนุพันธ์) หรือลดระดับของโดปามีน
Haloperidol (2–20 mg / วัน), tiaprid (100–600 mg / วัน) ไม่เกินสามเดือน, thioridazine (สูงถึง 100 mg / วัน), reserpine (0.25–2 mg / วัน), clonazepam (1–2 mg, 5-6 มก. / วัน) ยาเหล่านี้มีส่วนช่วยในการลดภาวะ hyperkinesis, ปรับความตึงเครียดทางอารมณ์, ชดเชยความผิดปกติทางบุคลิกภาพ
ในโรงพยาบาลการรักษาความผิดปกติทางจิตจะดำเนินการโดยคำนึงถึงกลุ่มอาการของโรคชั้นนำอายุและสภาพทั่วไปของผู้ป่วย ในการรักษาผู้ป่วยนอกหลักการของการรักษาจะเหมือนกัน (การบำรุงรักษาอย่างต่อเนื่องของความผิดปกติของการเคลื่อนไหวเปลี่ยนระยะของยาเสพติด) การใช้งานผู้ป่วยนอกของขนาดที่ต่ำกว่าของอินซูลิน
กิจกรรมการฟื้นฟูสมรรถภาพสำหรับภาวะสมองเสื่อมน้อยถึงปานกลาง ได้แก่ การบำบัดด้วยการจ้างงาน, การบำบัดทางจิตและการฝึกอบรมด้านความรู้ความเข้าใจ จำเป็นที่จะต้องทำงานกับสมาชิกในครอบครัวการสนับสนุนทางด้านจิตใจของคนที่ดูแลคนป่วย วิธีการหลักในการป้องกันโรคคือการให้คำปรึกษาด้านการแพทย์และพันธุกรรมของญาติสนิทของผู้ป่วยโดยมีการอ้างอิงไปยังการวิเคราะห์ DNA เพื่อตัดสินใจว่าจะให้กำเนิดหรือไม่
การพยากรณ์โรคโดยทั่วไปจะไม่เอื้อ หลักสูตรของโรคมีความก้าวหน้าช้า, โรคมักจะนำไปสู่ความตายใน 10-15 ปี
สิ่งที่รบกวนคุณ?
การรักษาโรคฮันติงตัน
การรักษาโรคฮันติงตันเป็นอาการ โรคลมชักและความวิตกกังวลสามารถระงับได้บางส่วนจากอินซูลิน (เช่น chlorpromazine 25-300 มก. รับประทาน 3 ครั้ง / วัน, haloperidol 5-45 มก. รับประทาน 2 ครั้ง / วัน) หรือ reserpine 0.1 มก. รับประทาน 1 ครั้ง / วัน ปริมาณจะเพิ่มขึ้นถึงระดับสูงสุดที่ยอมรับได้ (จนกว่าผลข้างเคียงจะปรากฏขึ้นเช่นอาการง่วงนอน, โรคพาร์กินสัน, สำหรับยา reserpine, ความดันเลือดต่ำ) เป้าหมายของการบำบัดเชิงประจักษ์คือการลดการส่งผ่านกลูตามาเทอจิคผ่านตัวรับ Nmethyl-O-aspartate และเพื่อสนับสนุนการผลิตพลังงานในไมโตคอนเดรีย การรักษาที่มุ่งเป้าไปที่การเพิ่ม GABA ในสมองนั้นไม่ได้ผล
การทดสอบทางพันธุกรรมและการให้คำปรึกษามีความสำคัญเนื่องจากอาการของโรคปรากฏตัวในช่วงปลายอายุคลอดบุตร บุคคลที่มีประวัติครอบครัวเป็นบวกและผู้ที่สนใจในการทดสอบจะถูกส่งไปยังศูนย์เฉพาะด้านโดยคำนึงถึงผลทางจริยธรรมและจิตวิทยาทั้งหมด
รักษาตามอาการของโรคฮันติงตัน
การรักษาที่มีประสิทธิภาพที่สามารถหยุดยั้งการลุกลามของโรคฮันติงตันยังไม่ได้รับการพัฒนา ทำการทดสอบยาต่าง ๆ ซ้ำ ๆ แต่ไม่สามารถรับผลกระทบที่สำคัญได้ Neuroleptics และคู่อริตัวรับโดปามีนอื่น ๆ ถูกนำมาใช้กันอย่างแพร่หลายเพื่อแก้ไขความผิดปกติทางจิตและการเคลื่อนไหวที่ไม่ตั้งใจในผู้ป่วยที่มีโรคฮันติงตัน การเคลื่อนไหวโดยไม่สมัครใจสะท้อนให้เห็นถึงความไม่สมดุลระหว่างระบบโดปามีนและระบบ GABAergic ดังนั้นยารักษาโรคจิตจะใช้ในการลดกิจกรรมโดปามีนส่วนเกิน อย่างไรก็ตามยาเหล่านี้เองอาจทำให้เกิดผลข้างเคียงทางปัญญาและ extrapyramidal เด่นชัด ยิ่งไปกว่านั้นยกเว้นกรณีเหล่านี้เมื่อผู้ป่วยพัฒนาโรคจิตหรือเร้าอารมณ์ประสิทธิภาพของพวกเขายังไม่ได้รับการพิสูจน์ Neuroleptics มักทำให้เกิดหรือซ้ำซ้อนกลืนลำบากหรือความผิดปกติของการเคลื่อนไหวอื่น ๆ Neuroleptics ของคนรุ่นใหม่เช่น risperidone, clozapine และ olanzapine อาจเป็นประโยชน์อย่างยิ่งในการรักษาโรคฮันติงตันเนื่องจากพวกมันทำให้เกิดผลข้างเคียงจากการ extrapyramidal ในระดับที่น้อยลง แต่สามารถลดอาการหวาดระแวงหวาดระแวงหรือเพิ่มความหงุดหงิด
Tetrabenazine และ reserpine ยังลดกิจกรรมของระบบ dopaminergic และสามารถลดความรุนแรงของการเคลื่อนไหวโดยไม่สมัครใจในระยะแรกของโรค อย่างไรก็ตามการแก้ไขเหล่านี้สามารถทำให้เกิดภาวะซึมเศร้า เนื่องจากโรคนี้มักทำให้เกิดภาวะซึมเศร้าผลข้างเคียงนี้จึง จำกัด การใช้ยา reserpine และ tetrabenazine ในช่วงปลายของโรคเซลล์ที่มีตัวรับสารโดปามีนจะตายดังนั้นประสิทธิภาพของตัวรับโดปามีนจะลดลงหรือหายไป
ผู้ป่วยที่มีอาการทางประสาท, ซึมเศร้า, และ Anxiolytics ใช้รักษาโรคจิต, ซึมเศร้าและหงุดหงิดง่ายในผู้ป่วยที่มีอาการของโรคฮันติงตัน แต่ควรกำหนดเฉพาะช่วงเวลาที่ผู้ป่วยมีอาการเหล่านี้ ยาเสพติดที่อาจเป็นประโยชน์ในระยะหนึ่งของโรคในขณะที่ดำเนินไปอาจกลายเป็นไม่ได้ผลหรือแม้กระทั่งมีผลข้างเคียง
ในผู้ป่วยโรคฮันติงตันได้ทำการทดสอบตัวรับ GABA เนื่องจากโรคฮันติงตันเปิดเผยว่าระดับ GABA ใน striatum ลดลงอย่างมีนัยสำคัญรวมถึงภาวะแพ้ไวของ GABA receptors ในเขตการฉายภาพ เบนโซได้พิสูจน์แล้วว่ามีประสิทธิภาพในกรณีที่การเคลื่อนไหวโดยไม่สมัครใจและความบกพร่องทางสติปัญญาเกิดขึ้นจากความเครียดและความวิตกกังวล ควรกำหนดขนาดยาที่ต่ำเหล่านี้เพื่อหลีกเลี่ยงความใจเย็น ในผู้ป่วยส่วนใหญ่ที่มีโรคฮันติงตันไม่มียาใดที่นำไปสู่การพัฒนาคุณภาพชีวิตอย่างมีนัยสำคัญ
เมื่อเริ่มมีอาการของโรคฮันติงตันในช่วงต้นซึ่งเกิดขึ้นกับอาการพาร์คินเนียนอาจพยายามใช้ยาโดปามินอจิก แต่ประสิทธิภาพของยามี จำกัด นอกจากนี้ levodopa สามารถก่อให้เกิดหรือเสริมสร้าง myoclonus ในผู้ป่วยเหล่านี้ ในเวลาเดียวกัน Baclofen สามารถลดความแข็งแกร่งในผู้ป่วยบางรายของโรคฮันติงตัน
การรักษาเชิงป้องกัน (ป้องกันระบบประสาท) ของโรคฮันติงตัน
แม้ว่าข้อบกพร่องทางพันธุกรรมในโรคฮันติงตันเป็นที่รู้จักกัน แต่ก็ยังไม่ชัดเจนว่ามันจะนำไปสู่การเสื่อมสภาพของเซลล์ประสาทที่เลือก เป็นที่เชื่อกันว่าการรักษาด้วยการป้องกันมีวัตถุประสงค์เพื่อลดความเครียดออกซิเดชันและผล excitotoxic อาจมีความสามารถในการชะลอหรือระงับการลุกลามของโรค สถานการณ์อาจคล้ายกับการเสื่อมสภาพของตับซึ่งข้อบกพร่องทางพันธุกรรมยังไม่เป็นที่ทราบมานานหลายปีอย่างไรก็ตามการรักษาเชิงป้องกันมีผลรอง - การสะสมของทองแดง - นำไปสู่ "การรักษา" ในเรื่องนี้สมมติฐานที่ว่าโรคฮันติงตันเกี่ยวข้องกับความผิดปกติของการเผาผลาญพลังงานและการตายของเซลล์เนื่องจากผล excitotoxic ดึงดูดความสนใจเป็นพิเศษ โรคนี้สามารถทำให้เซลล์ตายได้เนื่องจากการรวมตัวกันของ intranuclear ของชิ้นส่วน N-terminal ของโรคเกาต์รบกวนการทำงานของเซลล์และเมแทบอลิซึม กระบวนการนี้สามารถส่งผลกระทบต่อเซลล์ประสาทบางกลุ่มในระดับที่สูงกว่ากลุ่มอื่น ๆ เนื่องจากความไวที่สูงขึ้นของพวกเขาต่อความเสียหาย excitotoxic ในกรณีนี้การรักษาด้วยการป้องกันด้วยตัวรับกรดอะมิโน excitatory คู่อริหรือวิธีการป้องกันความเสียหายอนุมูลอิสระจะสามารถป้องกันหรือชะลอการโจมตีและความก้าวหน้าของโรค ในรูปแบบห้องปฏิบัติการของเส้นโลหิตตีบด้านข้าง amyotrophic จะได้รับการแสดงให้เห็นว่าสารต้านอนุมูลอิสระและคู่อริรับ (HAC) สามารถชะลอการลุกลามของโรค วิธีการที่คล้ายกันอาจมีประสิทธิผลในโรคฮันติงตัน ขณะนี้การทดลองทางคลินิกกำลังดำเนินการกับคู่ต่อสู้ตัวรับกลูตาเมตและตัวแทนที่เพิ่มฟังก์ชั่นของ II ที่ซับซ้อนของห่วงโซ่การขนส่งอิเล็กตรอนยล