ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
โรคอัชเชอร์
ตรวจสอบล่าสุด: 04.07.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
โรค Usher syndrome เป็นโรคทางพันธุกรรมที่มีอาการหูหนวกตั้งแต่กำเนิด และตาบอดลงเรื่อยๆ เมื่ออายุมากขึ้น การสูญเสียการมองเห็นมักเกี่ยวข้องกับโรคเรตินิติสพิกเมนโตซา ซึ่งเป็นกระบวนการเสื่อมของเม็ดสีในจอประสาทตา ผู้ป่วยโรค Usher syndrome จำนวนมากยังมีปัญหาด้านการทรงตัวอย่างรุนแรงอีกด้วย
ระบาดวิทยา
จากผลการศึกษาวิจัยพบว่ากลุ่มอาการอัชเชอร์ส่งผลกระทบต่อเด็กหูหนวก-ใบ้ที่เข้ารับการตรวจประมาณ 8% (การทดสอบดำเนินการในสถาบันเฉพาะสำหรับผู้ป่วยหูหนวก-ใบ้) พบโรคจอประสาทตาอักเสบจากเม็ดสีใน 6-10% ของผู้ป่วยที่เป็นโรคหูหนวกแต่กำเนิด ซึ่งพบในผู้ป่วยโรคจอประสาทตาอักเสบจากเม็ดสีประมาณ 30%
เชื่อกันว่าโรคนี้พบได้ประมาณ 3-10 คนจากประชากร 100,000 คนทั่วโลก โดยพบได้ทั้งในผู้หญิงและผู้ชายเท่าๆ กัน ประชากรโลกประมาณ 5-6% เป็นโรคนี้ ผู้ป่วยหูหนวกเรื้อรังในเด็กประมาณ 10% เกิดจากกลุ่มอาการ Usher ประเภท I และ II
ในสหรัฐอเมริกา ประเภท 1 และ 2 เป็นประเภทที่พบบ่อยที่สุด โดยรวมคิดเป็นประมาณ 90 ถึง 95 เปอร์เซ็นต์ของผู้ป่วยโรค Usher ทั้งหมดในเด็ก
สาเหตุ โรคอัชเชอร์
โรค Usher ชนิด I, II และ III มีสาเหตุทางพันธุกรรมแบบถ่ายทอดทางยีนด้อย ในขณะที่ชนิด IV ถือเป็นความผิดปกติของโครโมโซม X สาเหตุของอาการตาบอดและหูหนวกที่เกิดขึ้นจากโรคนี้ยังไม่ได้รับการศึกษาอย่างเพียงพอ สันนิษฐานว่าผู้ป่วยโรคนี้ไวต่อส่วนประกอบที่สามารถทำลายโครงสร้างของ DNA มากเกินไป นอกจากนี้ โรคนี้ยังอาจเกี่ยวข้องกับความผิดปกติของระบบภูมิคุ้มกัน แต่ในกรณีนี้ไม่มีภาพที่ชัดเจนของกระบวนการนี้
ในปี 1989 ได้มีการระบุความผิดปกติของโครโมโซมเป็นครั้งแรกในผู้ป่วยโรคประเภทที่ 2 ซึ่งอาจนำไปสู่วิธีการแยกยีนที่ทำให้เกิดโรคในอนาคต นอกจากนี้ ยังอาจสามารถระบุยีนเหล่านี้ในผู้ที่มีพาหะและพัฒนาวิธีทดสอบทางพันธุกรรมก่อนคลอดแบบพิเศษได้อีกด้วย
 [ 8 ]
[ 8 ]
ปัจจัยเสี่ยง
โรคนี้ถ่ายทอดทางพันธุกรรมเมื่อพ่อแม่เป็นโรคนี้ทั้งคู่ กล่าวคือ ถ่ายทอดจากยีนด้อย ลูกก็สามารถถ่ายทอดโรคนี้ได้เช่นกันหากพ่อแม่เป็นพาหะของยีนดังกล่าว หากพ่อแม่ในอนาคตมียีนดังกล่าว โอกาสที่ลูกจะมีโรคนี้คือ 1 ใน 4 คนที่มียีนเพียงยีนเดียวที่ทำให้เกิดโรคนี้ถือว่าเป็นพาหะ แต่ไม่มีอาการของโรคนี้ ในปัจจุบันยังไม่สามารถระบุได้ว่าบุคคลนั้นมียีนของโรคนี้หรือไม่
หากเด็กเกิดมาจากพ่อแม่ที่มีคนหนึ่งไม่มียีนดังกล่าว โอกาสที่เด็กจะถ่ายทอดโรคนี้จึงมีน้อยมาก แต่เขาจะเป็นพาหะแน่นอน
อาการ โรคอัชเชอร์
อาการของโรคอัชเชอร์ ได้แก่ การสูญเสียการได้ยินและการสะสมของเซลล์เม็ดสีที่ผิดปกติในโครงสร้างของดวงตา ผู้ป่วยจะเกิดภาวะเสื่อมของจอประสาทตา ซึ่งทำให้การมองเห็นลดลง และในกรณีรุนแรงที่สุดอาจสูญเสียการมองเห็นได้ในที่สุด
การสูญเสียการได้ยินจากประสาทรับเสียงอาจเป็นแบบเล็กน้อยหรือสมบูรณ์ และโดยปกติจะไม่ลุกลามตั้งแต่แรกเกิด อย่างไรก็ตาม โรคจอประสาทตาสีอาจเริ่มพัฒนาขึ้นในวัยเด็กหรือในภายหลัง ผลการทดสอบแสดงให้เห็นว่าสามารถคงความคมชัดของการมองเห็นส่วนกลางไว้ได้หลายปี แม้ว่าการมองเห็นรอบข้างจะแย่ลง (ภาวะที่เรียกว่า "การมองเห็นแบบอุโมงค์")
สิ่งเหล่านี้เป็นอาการหลักของโรค ซึ่งบางครั้งอาจมีความผิดปกติอื่นๆ ร่วมด้วย เช่น โรคจิตเภทและความผิดปกติทางจิตอื่นๆ ปัญหาเกี่ยวกับหูชั้นใน และ/หรือต้อกระจก
รูปแบบ
ระหว่างการวิจัย สามารถระบุโรคนี้ได้ 3 ประเภท รวมถึงโรคประเภทที่ 4 ซึ่งค่อนข้างหายาก
โรคประเภทที่ 1 มีลักษณะเด่นคือหูหนวกตั้งแต่กำเนิดและมีอาการทรงตัวผิดปกติ โดยเด็กเหล่านี้มักจะเริ่มเดินได้เมื่ออายุ 1.5 ปีเท่านั้น การมองเห็นเสื่อมลงมักเริ่มตั้งแต่อายุ 10 ขวบ และอาการตาบอดกลางคืนจะค่อยๆ พัฒนาไปเมื่ออายุ 20 ปี เด็กที่เป็นโรคประเภทนี้จะมีอาการการมองเห็นรอบข้างในเชิงลึกมากขึ้น
ในโรคประเภทที่ 2 มักมีอาการหูหนวกปานกลางหรือพิการแต่กำเนิด ในกรณีนี้ อาการหูหนวกบางส่วนมักจะไม่เกิดขึ้นอีก โรคจอประสาทตาอักเสบเป็นเม็ดสีจะเริ่มพัฒนาขึ้นเมื่อใกล้จะสิ้นสุดวัยรุ่นหรือหลังจากอายุ 20 ปี อาการตาบอดกลางคืนมักจะเริ่มเกิดขึ้นเมื่ออายุ 29-31 ปี การสูญเสียความสามารถในการมองเห็นในกรณีของโรคประเภทที่ 2 มักจะดำเนินไปช้ากว่าโรคประเภทที่ 1 เล็กน้อย
โรคประเภทที่ 3 มีลักษณะเฉพาะคือสูญเสียการได้ยินอย่างค่อยเป็นค่อยไป มักเริ่มในช่วงวัยแรกรุ่น และมีการพัฒนาขึ้นอย่างค่อยเป็นค่อยไปในช่วงเวลาเดียวกัน (ช้ากว่าการสูญเสียการได้ยินเล็กน้อย) ของโรคเรตินิติสพิกเมนโตซา ซึ่งอาจกลายเป็นปัจจัยหนึ่งในการพัฒนาอาการตาบอดอย่างค่อยเป็นค่อยไปได้
อาการแสดงของโรคประเภทที่ 4 มักเกิดขึ้นในผู้ชาย ในกรณีนี้จะพบอาการผิดปกติที่ค่อยๆ แย่ลง รวมถึงการสูญเสียการได้ยินและการมองเห็นด้วย โรคประเภทนี้พบได้น้อยมากและมักมีลักษณะเป็นโครโมโซม X
การวินิจฉัย โรคอัชเชอร์
การวินิจฉัยโรค Usher จะพิจารณาจากอาการหูหนวกเฉียบพลันและการสูญเสียการมองเห็นที่ค่อยๆ เป็นค่อยๆ ไปของผู้ป่วย
การทดสอบ
อาจมีคำสั่งให้ทดสอบทางพันธุกรรมพิเศษเพื่อตรวจหาการกลายพันธุ์
มีการค้นพบตำแหน่งทางพันธุกรรม 11 ตำแหน่งที่สามารถทำให้เกิดโรค Usher และมีการระบุยีน 9 ตำแหน่งที่เป็นสาเหตุแน่นอนของโรคนี้:
- ประเภท 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- ประเภทที่ 2: ush2a, VLGR1, WHRN
- กลุ่มอาการ Usher ประเภทที่ 3: USH3A
นักวิทยาศาสตร์จาก NIDCD ร่วมกับเพื่อนร่วมงานจากมหาวิทยาลัยในนิวยอร์กและอิสราเอล ได้ระบุการกลายพันธุ์ที่เรียกว่า R245X ในยีน Pcdh15 ซึ่งเป็นสาเหตุของโรค Usher ประเภท 1 ในประชากรชาวยิวจำนวนมาก
หากต้องการทราบข้อมูลเกี่ยวกับห้องปฏิบัติการที่ดำเนินการทดลองทางคลินิก โปรดไปที่ https://www.genetests.org และค้นหา "Usher syndrome" ในไดเร็กทอรีห้องปฏิบัติการ
หากต้องการเรียนรู้เกี่ยวกับการทดลองทางคลินิกที่มีอยู่ที่รวมถึงการทดสอบทางพันธุกรรมสำหรับโรค Usher โปรดไปที่ https://www.clinicaltrials.gov และค้นหา "โรค Usher" หรือ "การทดสอบทางพันธุกรรมของโรค Usher"
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
การวินิจฉัยเครื่องมือ
มีวิธีการวินิจฉัยเครื่องมือหลายวิธี:
- การตรวจดูบริเวณจอประสาทตาเพื่อตรวจหาจุดสีบนจอประสาทตา ตลอดจนการตีบแคบของหลอดเลือดในจอประสาทตา
- การตรวจคลื่นไฟฟ้าเรตินา ซึ่งช่วยตรวจจับความเบี่ยงเบนจากความเสื่อมเริ่มต้นในเรตินาของดวงตา โดยแสดงการสูญพันธุ์ของเส้นทางการตรวจคลื่นไฟฟ้าเรตินา
- อิเล็กโตรนีสแทกโมแกรม (ENG) จะวัดการเคลื่อนไหวของดวงตาโดยไม่ตั้งใจ ซึ่งอาจบ่งชี้ถึงการมีอยู่ของความไม่สมดุล
- การตรวจการได้ยินซึ่งใช้เพื่อตรวจหาการมีอยู่ของอาการหูหนวกและความรุนแรงของอาการ
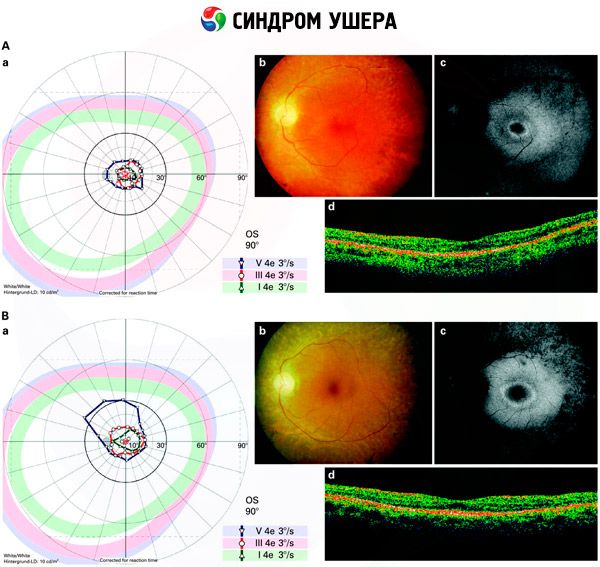
การวินิจฉัยที่แตกต่างกัน
โรค Usher จะต้องถูกแยกความแตกต่างจากโรคอื่นๆ ที่คล้ายคลึงกัน
กลุ่มอาการฮอลล์เกรน ซึ่งมีลักษณะเฉพาะคือสูญเสียการได้ยินแต่กำเนิดและสูญเสียการมองเห็นอย่างต่อเนื่อง (อาจเกิดต้อกระจกและตาสั่นได้) อาการเพิ่มเติม ได้แก่ อาการอะแท็กเซีย ความผิดปกติทางจิตและการเคลื่อนไหว โรคจิต และปัญญาอ่อน
โรคอัลสตรอม ซึ่งเป็นโรคทางพันธุกรรมที่จอประสาทตาเสื่อม ส่งผลให้สูญเสียการมองเห็นตรงกลาง โรคนี้มักเกิดขึ้นกับโรคอ้วนในวัยเด็ก ขณะเดียวกัน โรคเบาหวานและการสูญเสียการได้ยินจะเริ่มพัฒนาขึ้นหลังจากอายุ 10 ปี
โรคหัดเยอรมันในหญิงตั้งครรภ์ในช่วงไตรมาสแรกอาจทำให้เกิดความผิดปกติต่างๆ ในการพัฒนาการของทารกได้ ผลที่ตามมาของความผิดปกติดังกล่าว ได้แก่ การสูญเสียการได้ยิน รวมถึง (หรือ) ปัญหาทางสายตา และนอกจากนี้ยังมีข้อบกพร่องด้านพัฒนาการต่างๆ อีกด้วย
ใครจะติดต่อได้บ้าง?
การรักษา โรคอัชเชอร์
ปัจจุบันยังไม่มีวิธีรักษาโรค Usher syndrome ดังนั้นการบำบัดในกรณีนี้จึงประกอบด้วยการชะลอการสูญเสียการมองเห็นเป็นหลัก รวมถึงการชดเชยการสูญเสียการได้ยิน วิธีการรักษาที่เป็นไปได้ ได้แก่:
- การรับประทานวิตามินเอ (จักษุแพทย์บางคนเชื่อว่าวิตามินเอปาล์มิเตตในปริมาณสูงอาจช่วยชะลอการดำเนินของโรคเรตินิติสพิกเมนโตซาได้ แต่ไม่สามารถหยุดการดำเนินของโรคได้)
- การปลูกถ่ายอุปกรณ์อิเล็กทรอนิกส์พิเศษเข้าไปในหูของผู้ป่วย (เครื่องช่วยฟัง, ประสาทหูเทียม)
จักษุแพทย์แนะนำให้ผู้ใหญ่ส่วนใหญ่ที่เป็นโรคเรตินิติสพิกเมนโตซาชนิดทั่วไปรับประทานวิตามินเอปาล์มิเตต 15,000 IU (หน่วยสากล) ทุกวันภายใต้การดูแล เนื่องจากผู้ป่วยโรค Usher ชนิดที่ 1 ไม่รวมอยู่ในการศึกษา จึงไม่แนะนำให้ผู้ป่วยกลุ่มนี้รับประทานวิตามินเอในปริมาณสูง ผู้ที่กำลังพิจารณารับประทานวิตามินเอควรปรึกษากับแพทย์เกี่ยวกับทางเลือกในการรักษานี้ คำแนะนำอื่นๆ สำหรับทางเลือกในการรักษานี้ ได้แก่:
- การเปลี่ยนแปลงการบริโภคอาหารของคุณโดยรวมอาหารที่มีวิตามินเอสูง
- สตรีที่วางแผนจะตั้งครรภ์ควรหยุดรับประทานวิตามินเอในปริมาณสูงสามเดือนก่อนที่จะตั้งครรภ์เนื่องจากมีความเสี่ยงต่อการเกิดข้อบกพร่องทางการเกิดเพิ่มขึ้น
- สตรีมีครรภ์ควรหยุดรับประทานวิตามินเอในปริมาณสูง เนื่องจากมีความเสี่ยงต่อการเกิดข้อบกพร่องทางการเกิดเพิ่มขึ้น
การปรับเด็กให้เข้ากับชีวิตทางสังคมก็มีความสำคัญเช่นกัน ซึ่งต้องได้รับความช่วยเหลือจากครูการศึกษาพิเศษและนักจิตวิทยา ในกรณีที่ผู้ป่วยเริ่มมีอาการสูญเสียการมองเห็นมากขึ้น ควรสอนให้ใช้ภาษามือ
พยากรณ์
โรคอัชเชอร์มีการคาดการณ์ที่ไม่ดี การมองเห็นและความคมชัดของการมองเห็นจะเริ่มเสื่อมลงภายในระยะเวลา 20-30 ปีในผู้ป่วยส่วนใหญ่ที่เป็นโรคนี้ไม่ว่าประเภทใด ในบางกรณีอาจสูญเสียการมองเห็นทั้งสองข้างอย่างสมบูรณ์ การสูญเสียการได้ยินซึ่งมักจะมาพร้อมกับอาการใบ้จะพัฒนาอย่างรวดเร็วจนกลายเป็นสูญเสียการได้ยินทั้งสองข้างอย่างสมบูรณ์