ผู้เชี่ยวชาญทางการแพทย์ของบทความ

สิ่งตีพิมพ์ใหม่
แมงมุมนิ้ว
ตรวจสอบล่าสุด: 04.07.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
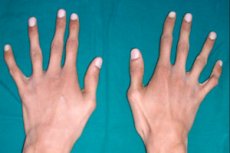
โรคเนื้อเยื่อเกี่ยวพันทางพันธุกรรมที่พบได้น้อยชนิดหนึ่ง คือ โรคอะแรคโนแดกทิลี ซึ่งเป็นความผิดปกติของนิ้ว ร่วมกับกระดูกท่อยาวขึ้น โครงกระดูกโค้งงอ มีความผิดปกติของระบบหัวใจและหลอดเลือดและอวัยวะที่การมองเห็น
โรคนี้ได้รับการอธิบายครั้งแรกโดยดร.วิลเลียมส์และกุมารแพทย์ชาวฝรั่งเศสชื่อมาร์แฟน ซึ่งเรียกโรคนี้ว่า dolichostenomelia ต่อมาคำว่ากลุ่มอาการมาร์แฟนปรากฏในวงการแพทย์ทั่วโลก และตั้งแต่ปี 1902 เป็นต้นมา คำว่า arachnodactyly ก็ถูกใช้กัน โรคนี้เกี่ยวข้องกับความผิดปกติแต่กำเนิดของเนื้อเยื่อเกี่ยวพัน และอาการสำคัญอย่างหนึ่งคือนิ้วที่ยาว บาง และโค้งงอเหมือนแมงมุม [ 1 ]
ระบาดวิทยา
โรค Arachnodactyly ถือเป็นโรคทางกรรมพันธุ์แบบโมโนเจนิกของเนื้อเยื่อเกี่ยวพัน โดยมีการถ่ายทอดทางออโตโซมัลโดมิแนนต์ ความสามารถในการซึมผ่านสูง และระดับการแสดงออกที่หลากหลาย
ในประมาณ 75-80% ของกรณี โรคนี้เป็นแบบถ่ายทอดทางพันธุกรรม และในกรณีที่เหลือ โรคนี้เกิดจากการกลายพันธุ์ตามธรรมชาติ (โดยเฉพาะอย่างยิ่ง เนื่องมาจากการกลายพันธุ์แบบ point missense) [ 2 ]
ได้รับการพิสูจน์แล้วว่าลักษณะทางพยาธิวิทยาของโรคอะแรคโนแดกทิลีเกิดจากการเปลี่ยนแปลงในยีนหลายชนิด (ใน 95% ของกรณี):
- ในยีนไฟบริลลิน-1 (มีการศึกษาการกลายพันธุ์ประมาณหนึ่งพันครั้ง) บนโครโมโซม 15q21.1
- ยีน TGFβR1 หรือ TGFβR2 บนโครโมโซม 9 และ 3p24.2-P25
การเปลี่ยนแปลงเหล่านี้ส่งผลโดยตรงต่อขอบเขตของอาการทางคลินิก
การกลายพันธุ์ในโซ่ α-2 ของคอลลาเจนชนิด I ได้รับการอธิบายไว้ในผู้ป่วย 5%
อุบัติการณ์ของโรคอะแรคโนแดกทิลีอยู่ที่ราว 2 รายต่อประชากร 5,000 คน โดยไม่มีการระบุเชื้อชาติหรือเพศ [ 3 ]
สาเหตุ แมงมุมนิ้ว
โรคนิ้วโป้งเกิดจากการกลายพันธุ์โดยธรรมชาติ โรคนี้ไม่ได้มีลักษณะเฉพาะของโรคทางพันธุกรรมแต่กำเนิดอย่างที่ทราบกันทั่วไป และนิ้วที่ยาวมักแสดงอาการในกลุ่มอาการมาร์แฟนและโฮโมซิสตินูเรียเป็นหลัก
โรคนี้พบได้น้อยและผู้ป่วยทั้งหมดที่พบมีการกลายพันธุ์ของยีน FBN1 (ยีนไฟบริลลิน) ซึ่งอยู่บนโครโมโซม 15 ในบริเวณ q21.1 ความหลากหลายทางคลินิกของโรคนี้สามารถอธิบายได้จากการกลายพันธุ์จำนวนมาก โดยประมาณ 15% เป็นการเปลี่ยนแปลงของการกลายพันธุ์ที่เกิดขึ้นระหว่างการปฏิสนธิ [ 4 ]
โรคอะแรคโนแดกทิลีชนิดผิดปกติเกิดจากการกลายพันธุ์ของยีนอื่น ๆ: มีการค้นพบความสัมพันธ์ระหว่างการพัฒนาของโรคและการกลายพันธุ์ของยีน LTBP3, R ซึ่งอยู่ในบริเวณโครโมโซม 14 ในบริเวณ q24 และแปลงปัจจัยการเจริญเติบโต
การกลายพันธุ์ส่วนใหญ่นั้นเรียกว่าการกลายพันธุ์แบบ missense (ประมาณ 57%) มากกว่า 20% เป็นการลบออกเล็กน้อย โดยมีการสูญเสียส่วนโครโมโซม ประมาณ 12% เป็นการกลายพันธุ์ที่ตำแหน่งการต่อยีน 8% เป็นการกลายพันธุ์แบบ non-ense และ 2% เป็นการจัดเรียงและการลบออกในระดับใหญ่ [ 5 ]
โรคนิ้วหดเกร็งแต่กำเนิดเกิดจากความผิดปกติในการสังเคราะห์โปรตีนที่ชื่อว่าไฟบริลลิน-2 ปัญหาเกิดจากการกลายพันธุ์ของยีน FBN2 ซึ่งอยู่บนโครโมโซมที่ 5 ในบริเวณ q23-q31 การกลายพันธุ์ทั้งหมดส่วนใหญ่เป็นการกลายพันธุ์แบบจุด โดยเปลี่ยนโคดอนเป็นรหัสของกรดอะมิโนตัวอื่น [ 6 ]
โฮโมซิสตินูเรียเกิดขึ้นจากการเผาผลาญกรดอะมิโนจำเป็นเมไทโอนีนที่บกพร่อง สาเหตุของโรคอาจมาจากการสูญเสียกิจกรรมหรือการลดลงของสารเอนไซม์ต่อไปนี้:
- เอนไซม์ซีสตาไธโอนีนเบตาซินเทส (CBS) โรคนี้ดื้อต่อวิตามินบี6และแสดงอาการด้วยอาการเฉพาะของรูปแบบที่ไม่ขึ้นกับวิตามินบี6อย่างไรก็ตาม การนำการเตรียมวิตามินบี6มาใช้ไม่ได้ทำให้อาการดีขึ้น
- เอนไซม์ N5, N10 เมทิลีนเทตระไฮโดรโฟเลตรีดักเตส (MTHFR) เอนไซม์นี้เป็นสารตั้งต้นที่ทำให้เกิดการดีเมทิลเลชันของโฮโมซิสเทอีนและเปลี่ยนเป็นเมไทโอนีน ซึ่งเป็นกรดอะมิโนจำเป็นที่เป็นส่วนประกอบของโปรตีนและสารเปปไทด์หลายชนิด
- เอนไซม์ N5 เมทิลีนเทตระไฮโดรโฟเลต เรากำลังพูดถึงรูปแบบของโรคที่ต้องอาศัยวิตามินบี6ซึ่งเกิดขึ้นจากการขาดวิตามินบี6การนำการเตรียมวิตามินเข้าสู่ร่างกายจะช่วยปรับปรุงสภาพของผู้ป่วยได้
- เอนไซม์โฮโมซิสเทอีนทรานส์เมทิลเลส พัฒนาขึ้นภายใต้สภาวะผิดปกติของการเผาผลาญของโคบาลามิน – วิตามินบี12
ปัจจัยเสี่ยง
โรคอะแรคนอดัคทิลีเป็นโรคทางพันธุกรรมที่มีลักษณะเฉพาะคือเนื้อเยื่อเกี่ยวพันได้รับความเสียหายอย่างเป็นระบบ พยาธิวิทยาเกี่ยวข้องกับการเปลี่ยนแปลงของยีนไฟบริลลิน 1 ซึ่งอยู่ที่แขนสั้นของโครโมโซม 15 ในตำแหน่ง 21.1
โรค Arachnodactyly ถ่ายทอดทางพันธุกรรมแบบออโตโซมัลโดมิแนนต์ โดยมีความสามารถในการแทรกซึมสูงและการแสดงออกที่หลากหลาย พยาธิสภาพนี้สามารถแสดงออกได้ทั้งในฝั่งชายและหญิง
กลไกการเกิดโรค
มากกว่าร้อยละ 50 ของมวลร่างกายมนุษย์ประกอบด้วยโครงสร้างเนื้อเยื่อเกี่ยวพัน โดยเฉพาะโครงกระดูก ผิวหนัง หลอดเลือด น้ำเหลือง และเลือด
เซลล์เนื้อเยื่อเกี่ยวพันแสดงโดยไฟโบรบลาสต์และชนิดย่อย ได้แก่ ออสเตียสบลาสต์ คอนโดรไซต์ เคอราโตบลาสต์ โอดอนโตบลาสต์ เช่นเดียวกับแมคโครฟาจและมาสต์เซลล์
เนื้อเยื่อเกี่ยวพันของตัวอ่อนเป็นวัสดุสำหรับการสร้างองค์ประกอบทางธรรมชาติ ทางพันธุกรรม และเอพิเจเนติกส์ โรคของเนื้อเยื่อเกี่ยวพันจะส่งผลต่อร่างกายโดยรวมในระดับหนึ่ง โดยขึ้นอยู่กับความสามารถในการทำงานและองค์ประกอบของร่างกาย
ในแมงมุมมีนิวคลีโอไทด์ที่แทนที่ยีนที่มีข้อมูลโครงสร้างเกี่ยวกับเปปไทด์ไฟบริลลิน-1 สารโปรตีนนี้จัดอยู่ในกลุ่มไกลโคโปรตีน มีส่วนร่วมในระบบไมโครไฟบริล และเป็นพื้นฐานของเส้นใยยืดหยุ่นของเนื้อเยื่อเกี่ยวพัน
เนื้อเยื่อรักษาโครงสร้างให้มั่นคงได้เนื่องจากเมทริกซ์ระหว่างเซลล์ซึ่งประกอบด้วยปัจจัยการเจริญเติบโตจำนวนมากที่ช่วยให้เซลล์เกิดการผลัดตัวใหม่ขึ้นอย่างเป็นระบบ หลอดเลือดและเอ็นขนาดใหญ่ประกอบด้วยเส้นใยอีลาสตินจำนวนมาก ซึ่งความเสียหายดังกล่าวทำให้เกิดอาการหลักของโรคอะแร็กโนแดกทิลี
ปัจจัยการเจริญเติบโตที่เปลี่ยนแปลง β ได้รับความเสียหายอย่างมาก: รูปแบบที่ไม่มีการใช้งานจะไม่ถูกจับ ซึ่งนำไปสู่การเพิ่มขึ้นของกิจกรรมทางชีวภาพของปัจจัยนี้
ความผิดปกติของเส้นใยทำให้เกิดการสร้างเส้นใยที่ผิดปกติ ส่งผลให้สูญเสียความแข็งแรงและความยืดหยุ่นของผิวหนังและโครงสร้างของเนื้อเยื่อเกี่ยวพันอื่นๆ
เมื่อโครงสร้างคอลลาเจนถูกทำลาย การเชื่อมโยงหลักในการหยุดเลือดก็จะถูกทำลาย
ผู้เชี่ยวชาญสังเกตเห็นว่าความไม่สมดุลของฮอร์โมนมีส่วนเกี่ยวข้องกับการเกิดและความก้าวหน้าของความผิดปกติในโครงสร้างของเนื้อเยื่อเกี่ยวพัน รวมถึงอาการของอแร็คโนแดกทิลี อาการของภาวะลิ่มเลือดอุดตันจะพิจารณาจากความผิดปกติทางรีโอโลยี โดยเฉพาะการเปลี่ยนแปลงความหนืดของเลือดในหลอดเลือดที่เปลี่ยนแปลงไปในบริเวณบราคิโอเซฟาลิก
ระบบย่อยอาหารได้รับผลกระทบจากความผิดปกติของคอลลาเจน: อาการเคลื่อนตัวของท่อน้ำดีผิดปกติ, ไส้เลื่อนบริเวณช่องเปิดของหลอดอาหารบริเวณกะบังลม, ความผิดปกติของตับและท่อน้ำดี, โรคกระเพาะและลำไส้อักเสบเรื้อรังแบบ "ลบออก" และภาวะ dolichosigma เป็นต้น
โรคอะแรคโนแดกทิลีถ่ายทอดได้อย่างไร?
โรค Arachnodactyly ถ่ายทอดทางพันธุกรรมเป็นลักษณะเด่นทางยีน แต่ก็สามารถเกิดขึ้นได้จากการกลายพันธุ์โดยธรรมชาติ กล่าวคือ พยาธิวิทยาไม่จำเป็นต้องแสดงอาการในญาติใกล้ชิดคนใดคนหนึ่ง เช่น ในพ่อแม่ ปู่ย่าตายาย การกลายพันธุ์ที่เป็นพื้นฐานของพยาธิวิทยาสามารถแสดงอาการออกมาได้โดยสุ่ม
อาการ แมงมุมนิ้ว
ในทางการแพทย์จะแยกความแตกต่างระหว่างอาการอะแร็กโนแด็กทิลีแบบแฝงและแบบเปิดเผย อาการอะแร็กโนแด็กทิลีแบบแฝงจะมีการเปลี่ยนแปลงเกิดขึ้น แต่จะเกิดขึ้นแยกกันและจำกัดอยู่ที่ระบบอวัยวะหนึ่งหรือสองระบบ อาการอะแร็กโนแด็กทิลีแบบเปิดเผยจะแสดงอาการออกมาเป็นความผิดปกติที่มองเห็นได้หลายอย่างซึ่งมีอาการรุนแรงแตกต่างกัน ในกรณีนี้ อาการของผู้ป่วยอาจค่อนข้างคงที่เป็นเวลาหลายสิบปีหรือค่อยๆ ดีขึ้น โดยแสดงอาการออกมาสัมพันธ์กับอวัยวะและระบบอื่นๆ
โดยทั่วไปสามารถตรวจพบโรคอะแรคโนแดกทิลีได้ภายในไม่กี่วันแรกหลังคลอดบุตร
สัญญาณภายนอกหลักของโรคอะแรคโนแดกทิลีคือนิ้วที่บางและยาว ซึ่งมีความโค้งตามแบบฉบับของนิ้วและมีขาคล้ายแมงมุมเนื่องจากมีเอ็นสั้น
นิ้วแมงมุมหรืออาการนิ้วโป้งจะมองเห็นได้ชัดเจนตั้งแต่ช่วงแรกเกิด แต่อาการจะชัดเจนขึ้นเมื่ออายุประมาณ 3 ขวบ
ตัวบ่งชี้การเจริญเติบโตยังเป็นลักษณะเฉพาะอีกด้วย โดยเด็กมักจะตัวสูง ปลายแขนและหน้าแข้งยาว แขนขาไม่สมส่วนและผอม ความไม่สมส่วนนี้เห็นได้ชัดโดยเฉพาะในบริเวณเอวและกระดูกสะบ้าที่สูง [ 7 ]
ความผิดปกติอื่น ๆ ของโครงกระดูก ได้แก่:
- กะโหลกศีรษะมีรูปร่างแคบและยาว (dolichocephalic) โดยมีบริเวณใบหน้าที่มีรูปร่างผิดปกติ
- รูปร่างของหน้าอกเป็นรูปกรวย (ที่เรียกว่า “นก”)
- กระดูกสันหลังโค้ง
- ข้อสะโพกเคลื่อนแต่กำเนิด
- การเคลื่อนของหัวเข่าและไหล่ที่เป็นนิสัย
- ความโค้งของกระดูกส้นเท้า;
- การก่อตัวของกระดูกงอก – การเจริญเติบโตของกระดูก
- เท้าแบน;
- การพัฒนาชั้นไขมันไม่เพียงพอ
อาการเพิ่มเติมของโรคอะแรคโนแดกทิลีอาจได้แก่:
- สายตาสั้น;
- ตาขาวออกสีน้ำเงิน
- การเคลื่อนของเลนส์
- ความผิดปกติของหัวใจและหลอดเลือด (โรคหัวใจ, หลอดเลือดแดงใหญ่โป่งพอง);
- ความผอมที่เด่นชัด;
- ข้อต่อมีการเคลื่อนไหวมากเกินไป
- ท้องฟ้าแบบ “โค้ง”
โรค Arachnodactyly มีลักษณะเด่นคือกระดูกท่อยาว ทำให้กระดูกโค้งงอหลายจุด นอกจากนี้ ผู้ป่วยหลายรายยังมีอาการผิดปกติของกล้ามเนื้อรูปพีระมิด การตอบสนองของเอ็นไม่สมมาตร ภาวะ anosocoria และ nystagmus ซึ่งเป็นผลจากพัฒนาการทางจิตใจที่ปกติ
หากเราพูดถึงอาการอะแรคโนแดกทิลีร่วมกับภาวะโฮโมซิสตินูเรีย ผู้ป่วยอาจมีอาการดังต่อไปนี้:
- อาการชักแบบเป็นระยะๆ
- ต้อหินทุติยภูมิที่มีเลนส์เคลื่อน
- จอประสาทตาหลุดลอก สายตาเอียง
- ความเสียหายต่อลำต้นหลอดเลือดแดง (รวมทั้งไต สมอง)
- โรคกระดูกพรุน;
- ภาวะลิ่มเลือดอุดตัน
- ความบกพร่องทางจิต
ความผิดปกติของนิ้วมือในทารกแรกเกิด
ในกรณีส่วนใหญ่ ผู้ป่วยสามารถตรวจพบอาการผิดปกติของนิ้วในเด็กได้ภายในไม่กี่วันแรกของชีวิต อย่างไรก็ตาม เมื่ออายุมากขึ้น อาการต่างๆ มักจะแย่ลง
สัญญาณแรกๆ ที่ทำให้สงสัยได้ว่าเป็นโรคนี้มักเป็นดังนี้:
- แขนขาและนิ้วที่ยาวผิดปกติ
- น้ำหนักตัวต่ำกว่าเกณฑ์แต่ร่างกายของเด็กยังอยู่ในสภาพที่เพียงพอ
- ใบหน้ายาว,ยาว;
- ไขมันในร่างกายไม่เพียงพอ, กล้ามเนื้อไม่พัฒนาเต็มที่;
- ความยืดหยุ่นของข้อต่อมากเกินไป
เมื่ออายุประมาณ 4 ขวบ ภาวะผิดปกติของนิ้วจะเริ่มมีตั้งแต่หน้าอก กระดูกสันหลังเริ่มโค้งงอ และมีเท้าแบน
ในส่วนของอวัยวะการมองเห็นนั้น พบว่ามีภาวะสายตาสั้น เลนส์ตาเหล่ โครงสร้างของกระจกตาที่เปลี่ยนแปลงไป ตาเหล่ กระบวนการสร้างภาพไม่ปกติในม่านตาและจอประสาทตา ความผิดปกติเหล่านี้สามารถสังเกตเห็นได้ตั้งแต่ 2-3 ปีแรกของชีวิตเด็ก แม้ว่าอาการจะค่อยๆ แย่ลงเมื่อเวลาผ่านไป
อันตรายที่ใหญ่ที่สุดเกิดจากความผิดปกติของระบบหัวใจและหลอดเลือด หากมีอาการผิดปกติดังกล่าว หากไม่มีการรักษาที่เหมาะสม ผู้ป่วยอาจเสียชีวิตตั้งแต่ในวัยเด็กได้ ในบรรดาโรคที่อันตรายเป็นพิเศษ อาจพบความเสียหายของผนังหลอดเลือด ความผิดปกติของหัวใจ ความผิดปกติของหลอดเลือดหัวใจ ในบางครั้ง เด็กที่มีอายุ 1 ปีแรก อาจตรวจพบภาวะหัวใจล้มเหลวได้
ความผิดปกติของนิ้วอาจมาพร้อมกับความเสียหายของอวัยวะอื่นๆ โดยเฉพาะระบบประสาท หลอดลมและปอด ผิวหนัง และระบบสืบพันธุ์และทางเดินปัสสาวะ ความผิดปกติเหล่านี้ตรวจพบได้ในระหว่างขั้นตอนการวินิจฉัย
ภาวะแทรกซ้อนและผลกระทบ
ผลข้างเคียงที่พบบ่อยที่สุดของโรคอะแรคโนแดกทิลีคือความเสียหายต่อหัวใจและระบบทางเดินหายใจ นอกจากนี้ ยังอาจเกิดปัญหากับการมองเห็นได้ ซึ่งอาจรวมถึงการสูญเสียการมองเห็นด้วย
ผู้ป่วยโรคอะแร็กโนแดกทิลีต้องได้รับการรักษาที่ซับซ้อนสำหรับโรคที่มีอยู่ทั้งหมด หากไม่ได้รับการรักษาดังกล่าว อายุขัยของผู้ป่วยจะลดลงอย่างมาก ผู้ป่วยบางรายอาจมีชีวิตอยู่ได้ถึง 40 ปีโดยไม่ได้รับการแทรกแซงทางการแพทย์ เมื่อเวลาผ่านไป ปัญหาของระบบหัวใจและหลอดเลือดจะแย่ลง ความเสี่ยงของการเกิดเลือดออกอย่างรุนแรงและผลลัพธ์ที่น่าเศร้าก็เพิ่มขึ้น
ผู้ที่เป็นโรคอะแร็กโนแดกทิลีควรได้รับการตรวจร่างกายเป็นประจำและรักษาโรคร่วมด้วย เนื่องจากโรคนี้เท่านั้นจึงจะมีโอกาสมีอายุยืนยาวโดยไม่มีภาวะแทรกซ้อนร้ายแรง
โรค Marfan และโรค arachnodactyly ต้องได้รับการดูแลเป็นพิเศษหากผู้ป่วยหญิงตั้งครรภ์ ในสถานการณ์เช่นนี้ สิ่งสำคัญคือต้องตรวจร่างกายผู้ป่วยเป็นประจำและละเอียดถี่ถ้วน รวมถึงต้องดูแลระบบหัวใจและหลอดเลือดของผู้ป่วยด้วย นี่เป็นวิธีเดียวที่จะรับประกันว่าการตั้งครรภ์จะประสบความสำเร็จและการคลอดบุตรอย่างปลอดภัย
เพื่อหลีกเลี่ยงผลข้างเคียง ผู้ป่วยโรคอะแรคโนแดกทิลีควรได้รับการตรวจจากแพทย์เป็นประจำเพื่อป้องกันการเกิดโรคเกี่ยวกับหลอดเลือดหัวใจและกล้ามเนื้อและโครงกระดูก แนะนำให้ผู้ป่วยดำเนินชีวิตอย่างมีสุขภาพดีและออกกำลังกายอย่างพอเหมาะ
การวินิจฉัย แมงมุมนิ้ว
การวินิจฉัยทางคลินิกสำหรับโรคอะแร็กโนแดกทิลีได้แก่ การเก็บรวบรวมอาการ ประวัติครอบครัว การประเมินลักษณะทางกายภาพ รวมถึงการตรวจร่างกายและการวัดส่วนต่างๆ ของร่างกาย สิ่งสำคัญคือต้องตรวจสอบการมีอยู่ของอาการแสดงของโรคเนื้อเยื่อเกี่ยวพันในญาติสายตรง เพื่อจุดประสงค์นี้ จึงมีการดำเนินการศึกษาทางลำดับวงศ์ตระกูล [ 8 ]
การวินิจฉัยทางห้องปฏิบัติการสำหรับโรคอะแร็กโนแดกทิลีเกี่ยวข้องกับการศึกษาสภาพของเนื้อเยื่อเกี่ยวพันบางประเภท เช่น เนื้อเยื่อกระดูกและกระดูกอ่อน เลือด และน้ำเหลือง การศึกษาระดับออกซีโพรลีนและไกลโคซามิโนไกลแคนในปัสสาวะประจำวันถือเป็นวิธีที่ให้ข้อมูลได้มากที่สุด นอกจากนี้ ยังต้องประเมินปริมาณไลซีน โพรลีน และออกซีโพรลีนในเลือดด้วย เพื่อตรวจสอบอัตราส่วนที่เปลี่ยนแปลงไปของคอลลาเจนประเภทต่างๆ และความผิดปกติของโครงสร้างของเส้นใยคอลลาเจน จะต้องกำหนดวิธีอิมมูโนฟลูออเรสเซนซ์ทางอ้อมโดยใช้แอนติบอดีโพลีโคลนัลต่อคอลลาเจนและไฟโบนิคติน [ 9 ] การทดสอบต่อไปนี้จะดำเนินการเพื่อสะท้อนคุณภาพของกระบวนการเผาผลาญของเนื้อเยื่อเกี่ยวพันโดยขึ้นอยู่กับข้อบ่งชี้:
- ไกลโคสะมิโนไกลแคน, ไฟบริลลิน, ไฟโบนิคติน;
- การศึกษาเกี่ยวกับไฮดรอกซีโพรลีน เครื่องหมายของการสังเคราะห์คอลลาเจนประเภทที่ 1 โพรเปปไทด์ปลายอะมิโนของโปรคอลลาเจนประเภทที่ 1 เครื่องหมายของการสลายคอลลาเจนประเภทที่ 1 กาแลกโตซิลออกซีไลซีน ดีออกซีไพริดินอลีน
- การวิเคราะห์การควบคุมการเผาผลาญคอลลาเจน (การศึกษาเมทริกซ์เมทัลโลโปรตีเนสและสารยับยั้งเนื้อเยื่อของเมทริกซ์เมทัลโลโปรตีเนส, ปัจจัยการเจริญเติบโตที่เปลี่ยนรูป);
- การวิเคราะห์ธาตุจุลภาคและมหภาค (ปริมาณแคลเซียมและแมกนีเซียม ฟอสฟอรัส เหล็ก กำมะถันและทองแดง โคบอลต์และซีลีเนียม สังกะสีและแมงกานีส ฟลูออรีนและวาเนเดียม ซิลิกอนและโบรอน)
- การศึกษาเครื่องหมายของการสร้างเนื้อเยื่อกระดูกและอัตราของกระบวนการสร้างแบบจำลองใหม่ (การวิเคราะห์เนื้อหาของออสเตโอแคลซิน ฟอสฟาเทสอัลคาไลน์ของกระดูก ฮอร์โมนพาราไทรอยด์ ฮอร์โมนโซมาโทโทรปิก โพรแลกติน วิตามิน ดี 3 เพ นโทซิดีน โฮโมซิสเทอีนในปัสสาวะและเลือด)
การวินิจฉัยด้วยเครื่องมือมีจุดมุ่งหมายเพื่อระบุข้อบกพร่องทางพัฒนาการที่มีอยู่และประเมินสถานะการทำงานของอวัยวะและระบบต่างๆ ความผิดปกติของหลอดเลือดหัวใจในโรคอะแรคโนแดกทิลีต้องรวมอยู่ในรายการวินิจฉัยของขั้นตอนต่างๆ เช่น:
- คลื่นไฟฟ้าหัวใจ;
- การตรวจวัดคลื่นไฟฟ้าหัวใจตลอด 24 ชั่วโมง;
- การตรวจอัลตราซาวด์หัวใจและหลอดเลือด
การตรวจความผิดปกติของหลอดเลือดจะทำโดยใช้การถ่ายภาพด้วยคอมพิวเตอร์หรือการถ่ายภาพด้วยคลื่นแม่เหล็กไฟฟ้า ขั้นตอนการวินิจฉัยเพิ่มเติมอาจรวมถึง:
- อัลตราซาวด์อวัยวะช่องท้อง ระบบสืบพันธุ์และระบบทางเดินปัสสาวะ;
- เอ็กซเรย์อวัยวะทรวงอก ข้อสะโพก; [ 10 ]
- CT หรือ MRI ของกระดูกสันหลัง
หลังจากการทดสอบที่จำเป็นทั้งหมดแล้ว ผู้ป่วยจะถูกส่งตัวไปรับคำปรึกษาทางพันธุกรรม
การวินิจฉัยที่แตกต่างกัน
กลุ่มอาการ Beals และอาการ arachnodactyly มีลักษณะคล้ายคลึงกันในกลุ่มอาการ Marfan (การกลายพันธุ์ของยีน FBN2 และ FBN1 ตามลำดับ) ซึ่งเกิดจากความเหมือนกันเกือบหมดของสารโปรตีน fibrillin 1 และ fibrillin 2 ไม่น่าแปลกใจที่ชื่อที่สองของกลุ่มอาการ Beals คือ contracture arachnodactyly [ 11 ]
การแยกความแตกต่างยังดำเนินการกับพยาธิสภาพของเนื้อเยื่อเกี่ยวพันอื่น ๆ ด้วย:
- โรคสติกเลอร์ซินโดรม;
- โรคเอห์เลอร์ส-ดันลอส
- โฮโมซิสตินูเรีย
- ข้อแข็งแข็ง
โรคบีลส์มีลักษณะเด่นคือมีรูปร่างคล้ายมาร์ฟานอยด์ มีการหดเกร็งแบบงอข้อใหญ่และข้อเล็กแต่กำเนิด มีอาการคล้ายแมงมุมที่ข้อมือและเท้า กระดูกสันหลังคด และใบหูมีรูปร่างผิดปกติ (เรียกว่าใบหู "พับ") การวิเคราะห์โมเลกุลของยีน FBN2 จะดำเนินการเพื่อยืนยันการวินิจฉัย
โรคข้อแข็งมีลักษณะเด่นคือข้อหดเกร็งหลายข้อ รูปร่างเตี้ย และสติปัญญาเสื่อม รูปร่างของนิ้วมือและหูไม่มีอะไรน่าสังเกต [ 12 ]
กลุ่มอาการเอห์เลอร์ส-ดานลอสมีลักษณะเด่นคือ กระดูกสันหลังคด กระดูกหน้าอกโค้งเป็นรูปกรวย เท้าแบนอย่างเห็นได้ชัด และการหักเหของแสงบกพร่อง การเคลื่อนไหวที่เพิ่มมากขึ้นและข้อต่อหลุดออก ผิวหนังมีความยืดหยุ่นและไวต่อความรู้สึกสูง ถือเป็นเรื่องปกติ
โรค Stickler มีลักษณะเฉพาะคือมีการเปลี่ยนแปลงของปริมาตรและความแข็งของข้อ
ใครจะติดต่อได้บ้าง?
การรักษา แมงมุมนิ้ว
โรค Arachnodactyly เป็นพยาธิวิทยาทางพันธุกรรม และปัจจุบันการแพทย์ยังไม่มีวิธีการแก้ไขข้อบกพร่องของยีน ดังนั้น การรักษาจึงมุ่งเป้าไปที่การปรับสภาพของผู้ป่วยให้เหมาะสมที่สุด ป้องกันไม่ให้พยาธิวิทยารุนแรงขึ้น และขจัดอาการที่แสดงออก แพทย์จะสั่งการรักษาที่ซับซ้อน โดยแพทย์ผู้เชี่ยวชาญหลายคนจะเข้าร่วมในคราวเดียวกัน ขึ้นอยู่กับประเภทของอาการที่เด่นชัดที่สุด โดยมักจะต้องติดต่อแพทย์กระดูกและข้อ แพทย์โรคหัวใจ แพทย์จักษุ แพทย์ทางเดินอาหาร และแพทย์เฉพาะทางอื่นๆ เพิ่มเติม [ 13 ]
ในคำแนะนำสำหรับการจัดการทางคลินิกของผู้ป่วย ถือเป็นหลักการทั่วไปดังต่อไปนี้:
- จำกัดกิจกรรมทางกาย แต่ไม่ถึงขั้นเลิกโดยสิ้นเชิง (เพื่อเป็นส่วนหนึ่งของการสนับสนุนระบบหัวใจและหลอดเลือด)
- การบำบัดด้วยยา;
- หากจำเป็น – การผ่าตัดแก้ไขบริเวณที่ได้รับผลกระทบมากที่สุดของหัวใจและหลอดเลือด
- การแก้ไขกระดูกและข้อ
- การบำบัดด้วยสปา, การกายภาพบำบัด, การออกกำลังกายเพื่อการบำบัด
อาหารของผู้ป่วยโรคอะแรคโนแดกทิลีควรประกอบด้วยอาหารที่มีโปรตีนสูง อุดมด้วยธาตุอาหาร วิตามิน และกรดไขมันในปริมาณที่เพียงพอ แนะนำให้รับประทานเนื้อสัตว์ ปลา อาหารทะเล ถั่ว และถั่วเปลือกแข็ง หากโรคอะแรคโนแดกทิลีเกิดจากโฮโมซิสตินูเรีย ควรจำกัดการบริโภคโปรตีนจากสัตว์อย่างมาก
แนะนำให้เด็กที่มีรูปร่างผอมและสูงเริ่มรับประทานน้ำมันเมล็ดฝ้าย น้ำมันถั่วเหลือง เมล็ดทานตะวัน ไขมันหมู และน้ำมันหมูตั้งแต่ยังเล็ก นอกจากนี้ ควรให้เด็กรับประทานผลิตภัณฑ์ที่มีกรดไขมันไม่อิ่มตัวเชิงซ้อน เช่น โอเมก้า ซึ่งจะไปยับยั้งการผลิตฮอร์โมนโซมาโตโทรปิก
เพื่อทำให้การเผาผลาญโปรตีนเป็นปกติ วิตามินกลุ่มบีจึงได้รับการกำหนดให้รับประทาน นอกจากนี้ยังสามารถได้รับจากผลิตภัณฑ์อาหาร เช่น บัควีท ถั่วลันเตา ตับ
กรดแอสคอร์บิกเข้าสู่ร่างกายของผู้ป่วยพร้อมกับอาหารเป็นสิ่งสำคัญมาก ดังนั้นการแช่โรสฮิป พริกหยวก กะหล่ำปลี ผลไม้รสเปรี้ยว รวมถึงซีบัคธอร์นและต้นหอมจึงรวมอยู่ในอาหาร
หากจำเป็น ผู้ป่วยจะได้รับการเสนอการแก้ไขกระดูกและข้อ ซึ่งจำเป็นในการลดภาระของกระดูกสันหลังและข้อต่อ โดยจะใช้รองเท้ากระดูก อุปกรณ์เข่าและอุปกรณ์อื่นๆ พื้นรองเท้า และผ้าพันแผลแบบยืดหยุ่น
การรักษาด้วยการผ่าตัดจะดำเนินการตามข้อบ่งชี้
ยา
การรักษาด้วยยาสำหรับโรคอะแรคโนแดกทิลีจะดำเนินการ 1-2 ครั้งต่อปี ขึ้นอยู่กับสภาพของผู้ป่วยและความรุนแรงของอาการทางพยาธิวิทยา ระยะเวลาของการรักษาจะกำหนดเป็นรายบุคคล โดยเฉลี่ยคือ 4 เดือน [ 14 ]
เพื่อกระตุ้นการสร้างคอลลาเจน จะมีการกำหนดให้ใช้ยาดังต่อไปนี้: Piaskledin 300, L-lysine หรือ L-proline ร่วมกับการเตรียมมัลติวิตามินที่ซับซ้อนที่ประกอบด้วยกรดแอสคอร์บิก วิตามินบี โทโคฟีรอล แมกนีเซียม สังกะสี ซีลีเนียม ทองแดง
ในกลุ่ม chondroprotectors การใช้ที่เหมาะสมที่สุดคือ chondroitin sulfate และ glucosamine sulfate ซึ่งเป็นยาที่มีส่วนร่วมในการควบคุมการเผาผลาญของเซลล์กระดูกอ่อน การยับยั้งการสังเคราะห์เอนไซม์ เพิ่มความไวของเซลล์กระดูกอ่อนต่อการทำงานของเอนไซม์ กระตุ้นกระบวนการสร้างเนื้อเยื่อ ฯลฯ ยาที่ใช้ร่วมกันถือเป็นยาที่เหมาะสมที่สุด เช่น Teraflex, Arthroflex, Arthra เป็นต้น
การเผาผลาญแร่ธาตุจะถูกกระตุ้นด้วยยาที่ทำให้กระบวนการฟอสฟอรัส-แคลเซียมเป็นปกติ ยาที่เลือกมักจะเป็นวิตามินดีรูปแบบที่ออกฤทธิ์: อัลฟาดี3 - Teva, Oxidevit, Bonviva เป็นต้น ในขณะเดียวกันก็ใช้การเตรียมฟอสฟอรัส แคลเซียม และแมกนีเซียม ในระหว่างการรักษา จะมีการตรวจระดับแคลเซียมและฟอสฟอรัสในเลือดหรือปัสสาวะประมาณ 20 วันครั้ง และจะทำการตรวจเลือดเพื่อหาฟอสฟาเตสด่าง
เพื่อปรับปรุงสถานะพลังงานของร่างกาย คุณสามารถกำหนด ฟอสฟาเดน, ริบอกซิน, เลซิติน, เอลการ์, โคเอนไซม์ Q10 ได้
รูปแบบการรักษาโดยประมาณอาจมีลักษณะดังนี้:
- รับประทานยาโคนโดรโพรเทคเตอร์ในปริมาณที่เหมาะสมกับวัย โดยรับประทานพร้อมอาหารและน้ำในปริมาณที่เพียงพอ ระยะเวลาในการรักษา 1 คอร์สคือ 3-4 เดือน
- แอล-โพรลีนในขนาด 500 มก. (สำหรับเด็กอายุ 12 ปีขึ้นไปและผู้ใหญ่) รับประทานก่อนอาหารครึ่งชั่วโมง วันละ 1-2 ครั้ง เป็นเวลา 6 สัปดาห์ หากจำเป็น อาจกำหนดให้รับประทานกรดอะมิโนคอมเพล็กซ์เพิ่มเติม ได้แก่ แอล-โพรลีน แอล-ไลซีน แอล-ลิวซีน ในปริมาณ 10-12 มก. ต่อน้ำหนักตัว 1 กก. รับประทานวันละ 1-2 ครั้ง เป็นเวลา 2 เดือน
- การเตรียมวิตามินและแร่ธาตุคอมเพล็กซ์ Centrum หรือ Vitrum หรือ Unicap โดยคำนึงถึงอายุ ระยะเวลาในการบริหารคือ 4 สัปดาห์
รูปแบบการรักษานี้เหมาะสมหากผู้ป่วยโรคอะแร็กโนแดกทิลียมีอาการปวดที่ระบบกล้ามเนื้อและโครงกระดูก และผลการทดสอบบ่งชี้ว่ามีการขับถ่ายไกลโคสะมิโนไกลแคนเพิ่มขึ้นในการทดสอบปัสสาวะประจำวัน และระดับกรดอะมิโนอิสระในเลือดลดลง
โดยทั่วไปแล้วผู้ป่วยจะรับการรักษาได้ดีโดยไม่มีผลข้างเคียงใดๆ หากตรวจพบอาการแพ้ยา แพทย์จะสั่งเปลี่ยนยาและปรับแผนการรักษา
การรักษาด้วยกายภาพบำบัด
แพทย์จะกำหนดขั้นตอนกายภาพบำบัดสำหรับโรคอะแรคโนแดกทิลีตามข้อบ่งชี้ ตัวอย่างเช่น ในกรณีที่กระดูกสร้างไม่เพียงพอ เพื่อปรับปรุงการสมานของกระดูกหัก หรือในกรณีที่มีสัญญาณของโรคกระดูกพรุน แนะนำให้ใช้อิเล็กโทรโฟรีซิสด้วยแคลเซียมคลอไรด์ 5% แมกนีเซียมซัลเฟต 4% คอปเปอร์ซัลเฟต 2% หรือสังกะสีซัลเฟต 2%
หากตรวจพบความผิดปกติของระบบหลอดเลือด ให้ใช้โซเดียมคาเฟอีนเบนโซเอต 1% เอเฟดรีนไฮโดรคลอไรด์ หรือเมซาตอน
เพื่อกระตุ้นการทำงานของเปลือกต่อมหมวกไต แพทย์จะสั่งให้ทำการรักษาด้วยอิเล็กโทรโฟรีซิสด้วยเอทิมิโซล 1.5% และการบำบัดด้วยเดซิเมตรที่บริเวณต่อมหมวกไต [ 15 ]
เพื่อรักษาสมดุลของหลอดเลือด แนะนำให้ใช้วิธีทางน้ำที่ส่งเสริม "ยิมนาสติก" ของหลอดเลือด การอาบน้ำ เช่น คาร์บอนไดออกไซด์ ไพน์ คลอไรด์-ไฮโดรเจน ไฮโดรเจนซัลไฟด์ และเรดอน เป็นวิธีที่ดีเยี่ยม การนวด การราดน้ำ การอาบน้ำแบบสลับสี การอาบน้ำแบบโฟมและเกลือ
เพื่อปรับปรุงสภาพเนื้อเยื่อกระดูกอ่อน จะมีการใช้การบำบัดด้วยแม่เหล็ก การบำบัดด้วยการเหนี่ยวนำ การบำบัดด้วยเลเซอร์ และการวิเคราะห์ด้วยอิเล็กโทรโฟรีซิสด้วยไดเมทิลซัลฟอกไซด์ (Dimexide)
การรักษาด้วยการผ่าตัด
การผ่าตัดเพื่อรักษาภาวะนิ้วโป้งถูกกำหนดให้เป็นไปตามข้อบ่งชี้อย่างเคร่งครัด ตัวอย่างเช่น หากตรวจพบความผิดปกติทางการไหลเวียนเลือดอย่างรุนแรงโดยมีลิ้นหัวใจยื่นออกมา หลอดเลือดแดงใหญ่โป่งพองอย่างรุนแรง ต้องเปลี่ยนลิ้นหัวใจ และส่วนหลอดเลือดแดงใหญ่ได้รับความเสียหาย [ 16 ]
หากมีอาการผิดปกติทางการทำงานของระบบทางเดินหายใจและระบบหัวใจและหลอดเลือดที่เด่นชัดซึ่งเกิดจากความโค้งของหน้าอกอย่างรุนแรง จะต้องผ่าตัดศัลยกรรมทรวงอก
ในกรณีของอาการปวดที่ค่อยๆ ลุกลามจากโรคกระดูกสันหลังคดอย่างรุนแรง 3-4 องศา การผ่าตัดก็เป็นสิ่งที่จำเป็นเช่นกัน จากมุมมองของจักษุวิทยา ข้อบ่งชี้ที่ชัดเจนในการเอาเลนส์ออก ได้แก่ การเคลื่อนของเลนส์ซึ่งเกิดจากต้อหินทุติยภูมิร่วมด้วย ตลอดจนต้อกระจกและการเปลี่ยนแปลงของจอประสาทตาที่เสื่อมลงซึ่งมีความเสี่ยงสูงที่จะหลุดลอก
การผ่าตัดใดๆ กับผู้ป่วยที่มีอาการผิดปกติของเส้นประสาทและโรคอื่นๆ ที่ส่งผลต่อโครงสร้างของเนื้อเยื่อเกี่ยวพัน จะทำเฉพาะในระยะที่อาการทางคลินิกและทางชีวเคมีดีขึ้นเท่านั้น หลังจากการผ่าตัด จำเป็นต้องติดตามอาการทางการแพทย์ในระยะยาวและการบำบัดอย่างเข้มข้นด้วยยาที่ปรับปรุงการเผาผลาญของเนื้อเยื่อเกี่ยวพัน
การป้องกัน
โรค Arachnodactyly เป็นความผิดปกติทางพันธุกรรมที่พบได้ยาก โดยบางครั้งอาจเกิดขึ้นเองโดยไม่ได้ตั้งใจในพ่อแม่ที่ดูเหมือนจะแข็งแรงดี ดังนั้น การป้องกันโรคล่วงหน้าจึงเป็นเรื่องยากมาก
หากผู้ปกครองคนใดคนหนึ่งมีประวัติครอบครัวที่ไม่ดี นั่นคือ ทราบว่ามีคนในครอบครัวเป็นโรคอะแร็กโนแดกทิลี คู่สมรสควรไปพบนักพันธุศาสตร์และทำการตรวจทางพันธุกรรมในขั้นตอนการวางแผนมีบุตร หลังจากยืนยันการตั้งครรภ์แล้ว แพทย์จะทำการวินิจฉัยก่อนคลอด รวมถึงการตรวจอัลตราซาวนด์ของทารกในครรภ์และการทดสอบทางชีวเคมีหลายอย่าง:
- การตรวจเลือดมารดา;
- การวิเคราะห์น้ำคร่ำ;
- การสุ่มตัวอย่างเนื้อเยื่อรกแกะ
- การวิเคราะห์เซลล์รกและเลือดจากสายสะดือ
ผู้ป่วยโรคอะแร็กโนแดกทิลีจะต้องอยู่ภายใต้การดูแลของแพทย์ตลอดชีวิต การป้องกันในผู้ป่วยมีจุดมุ่งหมายเพื่อป้องกันภาวะแทรกซ้อน โดยจะทำการผ่าตัดแก้ไขหัวใจ จ่ายยาเพื่อขจัดความเสี่ยงของการเกิดลิ่มเลือด จ่ายยาปฏิชีวนะเป็นระยะๆ
พยากรณ์
การพยากรณ์โรคสำหรับชีวิตของผู้ป่วยโรคอะแร็กโนแดกทิลีนั้นขึ้นอยู่กับความรุนแรงของอาการผิดปกติที่เกิดขึ้นเป็นหลัก โดยเฉพาะอย่างยิ่งโรคของระบบหัวใจและหลอดเลือด ระบบกล้ามเนื้อและโครงกระดูก และอวัยวะที่เกี่ยวข้องกับการมองเห็น โดยส่วนใหญ่แล้วพยาธิวิทยาจะซับซ้อนด้วยการแตกและแยกของหลอดเลือดแดงใหญ่ ดังนั้นจึงต้องคำนึงถึงเรื่องนี้และต้องทำการผ่าตัดหัวใจเพื่อแก้ไขอย่างทันท่วงที ซึ่งจะช่วยยืดอายุผู้ป่วยและปรับปรุงคุณภาพชีวิตของผู้ป่วย
หากวินิจฉัยได้ทันท่วงทีและปฏิบัติตามคำแนะนำของแพทย์ การพยากรณ์โรคก็ถือว่าดีในระดับหนึ่ง การช่วยเหลือทางการแพทย์ช่วยบรรเทาอาการของโรคนี้ลงได้อย่างมาก และผู้ป่วยก็มีโอกาสใช้ชีวิตได้ตามปกติและค่อนข้างสมบูรณ์โดยไม่มีภาวะแทรกซ้อนร้ายแรง