สิ่งตีพิมพ์ใหม่
ค้นพบโปรตีนสำคัญเพื่อป้องกันการสูญเสียมวลกระดูกในโรคกระดูกพรุน
ตรวจสอบล่าสุด: 02.07.2025

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
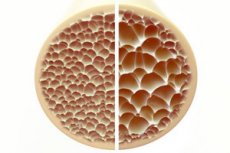
โรคกระดูกพรุนเป็นภาวะที่กระดูกมีรูพรุนและเปราะบาง เป็นอันตรายต่อสุขภาพกระดูก กระดูกเป็นโครงสร้างหลักที่คอยพยุงร่างกายของมนุษย์และคอยพยุงร่างกายให้ดำรงอยู่ต่อไป เมื่อมวลกระดูกลดลง ไม่เพียงแต่จะทำให้การพยุงร่างกายลดลงเท่านั้น แต่ยังทำให้การทำงานโดยรวมลดลงด้วย ส่งผลให้คุณภาพชีวิตลดลง
เนื่องจากอุบัติการณ์ของโรคกระดูกพรุนเพิ่มขึ้นในกลุ่มผู้สูงอายุ ทำให้ทรัพยากรด้านการดูแลสุขภาพสำหรับการดูแลระยะยาวมีภาระเพิ่มขึ้น ดังนั้น จึงจำเป็นต้องทำความเข้าใจกลไกที่ส่งผลต่อการเกิดโรคกระดูกพรุน และพัฒนาวิธีการรักษาแบบตรงจุดที่มีประสิทธิผลเพื่อลดผลกระทบในระยะยาว
เซลล์สร้างกระดูกและเซลล์สลายกระดูกเป็นเซลล์ 2 ประเภทที่มีบทบาทสำคัญในการบำรุงรักษาและปรับเปลี่ยนเนื้อเยื่อกระดูก ในขณะที่เซลล์สร้างกระดูกเป็นเซลล์ที่สร้างกระดูกซึ่งมีหน้าที่ในการสังเคราะห์และสะสมเนื้อเยื่อกระดูกใหม่ เซลล์สลายกระดูกเป็นเซลล์ที่ทำลายกระดูกซึ่งมีส่วนเกี่ยวข้องในการสลายและกำจัดเนื้อเยื่อกระดูกเก่าหรือเสียหาย
การเพิ่มขึ้นของสัดส่วนของกระดูกอ่อนนำไปสู่การสูญเสียมวลกระดูกในภาวะต่างๆ เช่น โรคกระดูกพรุน โรคข้ออักเสบรูมาตอยด์ (ข้ออักเสบ) และการแพร่กระจายของมะเร็งไปที่กระดูก กระดูกอ่อนเกิดจากการแยกตัวของแมคโครฟาจหรือโมโนไซต์ ซึ่งเป็นเซลล์ภูมิคุ้มกันชนิดหนึ่ง
ดังนั้น การยับยั้งการแบ่งตัวของกระดูกอ่อนอาจใช้เป็นกลยุทธ์การรักษาเพื่อป้องกันการสูญเสียมวลกระดูก อย่างไรก็ตาม กลไกโมเลกุลที่ชัดเจนที่ควบคุมกระบวนการสร้างกระดูกที่ซับซ้อนยังคงไม่ชัดเจน
ในการศึกษาวิจัยใหม่นี้ ศาสตราจารย์ทาดาโยชิ ฮายาตะ คุณทาคุโตะ คอนโนะ และคุณฮิโตมิ มูราจิ จากมหาวิทยาลัยวิทยาศาสตร์โตเกียว พร้อมด้วยเพื่อนร่วมงาน ได้ศึกษาวิจัยเกี่ยวกับการควบคุมระดับโมเลกุลของการแบ่งตัวของเซลล์สลายกระดูก การกระตุ้นด้วย RANKL ซึ่งเป็นตัวกระตุ้นตัวรับของลิแกนด์แฟกเตอร์นิวเคลียร์แคปปาบี (receptor activator of nuclear factor kappa B ligand) จะทำให้แมคโครฟาจเกิดการแบ่งตัวเป็นเซลล์สลายกระดูก
นอกจากนี้ โปรตีนสร้างกระดูก (BMP) และเส้นทางการส่งสัญญาณของทรานส์ฟอร์มิงโกรทแฟกเตอร์ (TGF)-β ยังเกี่ยวข้องกับการควบคุมการแบ่งตัวของเซลล์กระดูกอ่อนที่เกิดจาก RANKL ในการศึกษาปัจจุบันนี้ นักวิจัยมุ่งหวังที่จะศึกษาบทบาทของ Ctdnep1 ซึ่งเป็นฟอสฟาเทส (เอนไซม์ที่กำจัดกลุ่มฟอสเฟต) ซึ่งมีรายงานว่าสามารถยับยั้งเส้นทางการส่งสัญญาณของ BMP และ TGF-β ได้
การศึกษานี้ได้รับการตีพิมพ์ในวารสาร Biochemical and Biophysical Research Communications
ศาสตราจารย์ฮายาตะกล่าวว่า "RANKL ทำหน้าที่เป็น 'ตัวเร่ง' สำหรับการแบ่งตัวของเซลล์กระดูกอ่อน การขับรถต้องใช้ทั้งตัวเร่งและเบรกด้วย จากการทดลองนี้ เราพบว่า Ctdnep1 ทำหน้าที่เป็น 'เบรก' ในกระบวนการแบ่งตัวของเซลล์กระดูกอ่อน"
นักวิจัยได้ตรวจสอบการแสดงออกของ Ctdnep1 ในเซลล์แมคโครฟาจของหนูที่ได้รับการรักษาด้วย RANKL และเซลล์ควบคุมที่ไม่ได้รับการรักษาเป็นอันดับแรก พวกเขาสังเกตว่าการแสดงออกของ Ctdnep1 ไม่เปลี่ยนแปลงเมื่อตอบสนองต่อการกระตุ้นของ RANKL อย่างไรก็ตาม การแสดงออกของ Ctdnep1 จะอยู่เฉพาะในไซโตพลาซึมในรูปแบบเม็ดในแมคโครฟาจ และจะแยกความแตกต่างเป็นเซลล์สลายกระดูก ซึ่งแตกต่างจากตำแหน่งปกติรอบนิวเคลียสในเซลล์ประเภทอื่น ซึ่งบ่งชี้ถึงหน้าที่ในไซโตพลาซึมของ Ctdnep1 ในกระบวนการแยกความแตกต่างของเซลล์สลายกระดูก
นอกจากนี้ การน็อคดาวน์ของ Ctdnep1 (การลดการแสดงออกของยีน) ส่งผลให้จำนวนของกระดูกอ่อนที่เป็นบวกต่อฟอสฟาเทสกรดดื้อต่อทาร์เตรต (TRAP) เพิ่มขึ้น โดยที่ TRAP เป็นเครื่องหมายของกระดูกอ่อนที่แตกต่างกัน
การน็อกเอาต์ Ctdnep1 ส่งผลให้มีการแสดงออกของเครื่องหมายการแยกความแตกต่างที่สำคัญเพิ่มขึ้น รวมทั้ง "Nfatc1" ซึ่งเป็นปัจจัยการถอดรหัสหลักที่เหนี่ยวนำโดย RANKL สำหรับการแยกความแตกต่างของกระดูกอ่อน ผลลัพธ์เหล่านี้สนับสนุน "ฟังก์ชันเบรก" ของ Ctdnep1 ซึ่งควบคุมการแยกความแตกต่างของกระดูกอ่อนในทางลบ นอกจากนี้ การน็อกเอาต์ Ctdnep1 ยังส่งผลให้มีการดูดซึมแคลเซียมฟอสเฟตเพิ่มขึ้น ซึ่งบ่งชี้ว่า Ctdnep1 มีบทบาทยับยั้งในการสลายตัวของกระดูก
ในที่สุด แม้ว่าการน็อกเอาต์ Ctdnep1 จะไม่เปลี่ยนแปลงการส่งสัญญาณ BMP และ TGF-β แต่เซลล์ที่ขาด Ctdnep1 ก็แสดงระดับโปรตีนที่ถูกฟอสโฟรีเลต (เปิดใช้งานแล้ว) ที่เพิ่มขึ้น ซึ่งเป็นผลิตภัณฑ์ของเส้นทางการส่งสัญญาณ RANKL ผลลัพธ์เหล่านี้บ่งชี้ว่าผลการยับยั้งของ Ctdnep1 ต่อการแบ่งตัวของเซลล์กระดูกอ่อนอาจไม่ได้เกิดขึ้นผ่านการส่งสัญญาณ BMP และ TGF-β แต่เกิดขึ้นผ่านการลดระดับของเส้นทางการส่งสัญญาณ RANKL และระดับโปรตีน Nfatc1
โดยรวมแล้ว ผลลัพธ์เหล่านี้ให้ข้อมูลเชิงลึกใหม่ๆ เกี่ยวกับกระบวนการสร้างกระดูกอ่อนและระบุเป้าหมายการรักษาที่มีศักยภาพที่สามารถใช้ในการพัฒนาวิธีการรักษาเพื่อลดการสูญเสียมวลกระดูกอันเนื่องมาจากการทำงานของกระดูกอ่อนมากเกินไป นอกจากโรคที่มีลักษณะการสูญเสียมวลกระดูกแล้ว Ctdnep1 ยังได้รับการระบุว่าเป็นปัจจัยที่ทำให้เกิด medulloblastoma ซึ่งเป็นเนื้องอกในสมองในวัยเด็กอีกด้วย ผู้เขียนมีความหวังว่าการวิจัยของพวกเขาสามารถขยายไปยังโรคอื่นๆ ในมนุษย์ได้นอกเหนือจากการเผาผลาญของกระดูก
ศาสตราจารย์ฮายาตะสรุปว่า “ผลลัพธ์ของเราชี้ให้เห็นว่าจำเป็นต้องมี Ctdnep1 เพื่อป้องกันการเกิดกระดูกสลายมากเกินไป ผลลัพธ์เหล่านี้อาจขยายความรู้ของเราเกี่ยวกับวิธีที่เครือข่ายฟอสโฟรีเลชั่น-ดีฟอสโฟรีเลชั่นควบคุมการแบ่งตัวของกระดูกสลาย และอาจให้แนวทางการรักษาใหม่ๆ สำหรับการรักษาโรคกระดูกที่เกี่ยวข้องกับกิจกรรมของกระดูกสลายมากเกินไป”