โปรตีนสำคัญที่ระบุในการป้องกันการสูญเสียมวลกระดูกในโรคกระดูกพรุน
ตรวจสอบล่าสุด: 14.06.2024

เนื้อหา iLive ทั้งหมดได้รับการตรวจสอบทางการแพทย์หรือตรวจสอบข้อเท็จจริงเพื่อให้แน่ใจว่ามีความถูกต้องตามจริงมากที่สุดเท่าที่จะเป็นไปได้
เรามีแนวทางการจัดหาที่เข้มงวดและมีการเชื่อมโยงไปยังเว็บไซต์สื่อที่มีชื่อเสียงสถาบันการวิจัยทางวิชาการและเมื่อใดก็ตามที่เป็นไปได้ โปรดทราบว่าตัวเลขในวงเล็บ ([1], [2], ฯลฯ ) เป็นลิงก์ที่คลิกได้เพื่อการศึกษาเหล่านี้
หากคุณรู้สึกว่าเนื้อหาใด ๆ ของเราไม่ถูกต้องล้าสมัยหรือมีข้อสงสัยอื่น ๆ โปรดเลือกแล้วกด Ctrl + Enter
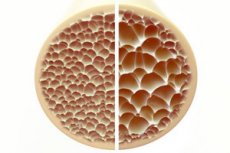
โรคกระดูกพรุน ซึ่งเป็นภาวะที่มีลักษณะของกระดูกที่มีรูพรุนและเปราะ ถือเป็นภัยคุกคามที่สำคัญต่อสุขภาพของโครงกระดูก กระดูกซึ่งเป็นโครงสร้างหลักของร่างกายมนุษย์ให้การสนับสนุนที่สำคัญ เมื่อมวลกระดูกลดลง ไม่เพียงแต่ทำให้การรองรับนี้ลดลงเท่านั้น แต่ยังทำให้การทำงานโดยรวมลดลงด้วย ส่งผลให้คุณภาพชีวิตลดลง
ในขณะที่อุบัติการณ์ของโรคกระดูกพรุนเพิ่มขึ้นในประชากรสูงวัย ความเครียดในทรัพยากรด้านการดูแลสุขภาพสำหรับการดูแลระยะยาวก็เพิ่มขึ้น ดังนั้นจึงจำเป็นต้องเข้าใจกลไกที่มีส่วนทำให้เกิดโรคกระดูกพรุน และพัฒนาวิธีการรักษาแบบกำหนดเป้าหมายที่มีประสิทธิผลเพื่อลดผลกระทบในระยะยาว
เซลล์สร้างกระดูกและเซลล์สร้างกระดูกเป็นเซลล์สองประเภทที่มีบทบาทสำคัญในการบำรุงรักษาและการเปลี่ยนแปลงเนื้อเยื่อกระดูก ในขณะที่เซลล์สร้างกระดูกเป็นเซลล์ที่สร้างกระดูกและมีหน้าที่ในการสังเคราะห์และการสะสมของเนื้อเยื่อกระดูกใหม่ แต่เซลล์สร้างกระดูกเป็นเซลล์ที่ย่อยสลายกระดูกซึ่งเกี่ยวข้องกับการย่อยสลายและการกำจัดเนื้อเยื่อกระดูกเก่าหรือที่เสียหาย
การเพิ่มสัดส่วนของเซลล์สร้างกระดูกทำให้เกิดการสูญเสียกระดูกในสภาวะต่างๆ เช่น โรคกระดูกพรุน โรคข้ออักเสบรูมาตอยด์ (การอักเสบของข้อต่อ) และการแพร่กระจายของกระดูก (มะเร็งที่แพร่กระจายไปยังกระดูก) เซลล์สร้างกระดูกเกิดขึ้นจากความแตกต่างของมาโครฟาจหรือโมโนไซต์ ซึ่งเป็นเซลล์ภูมิคุ้มกันประเภทหนึ่ง
การยับยั้งการสร้างความแตกต่างของเซลล์สร้างกระดูกจึงอาจใช้เป็นกลยุทธ์ในการรักษาเพื่อป้องกันการสูญเสียมวลกระดูก อย่างไรก็ตาม กลไกระดับโมเลกุลที่แม่นยำซึ่งควบคุมกระบวนการที่ซับซ้อนของการเปลี่ยนแปลงกระดูกยังคงไม่ชัดเจน
ในการศึกษาใหม่ ศาสตราจารย์ทาดาโยชิ ฮายาตะ นายทาคุโตะ คอนโนะ และนางสาวฮิโตมิ มูราจิ จากมหาวิทยาลัยวิทยาศาสตร์โตเกียว พร้อมด้วยเพื่อนร่วมงาน ได้เจาะลึกยิ่งขึ้นในการควบคุมระดับโมเลกุลของการสร้างความแตกต่างของเซลล์สร้างกระดูก การกระตุ้นโดยลิแกนด์กระตุ้นแคปปา B ของตัวรับนิวเคลียร์ (RANKL) กระตุ้นให้เกิดการเปลี่ยนแปลงของมาโครฟาจไปเป็นเซลล์สร้างกระดูก
นอกจากนี้ โปรตีน morphogenetic ของกระดูก (BMP) และการเปลี่ยนปัจจัยการเจริญเติบโต (TGF) - เส้นทางการส่งสัญญาณ β มีส่วนเกี่ยวข้องในการควบคุมการสร้างความแตกต่างของเซลล์สร้างกระดูกที่พึ่ง RANKL ในการศึกษาในปัจจุบัน นักวิจัยพยายามตรวจสอบบทบาทของ Ctdnep1 ซึ่งเป็นฟอสฟาเตส (เอนไซม์ที่กำจัดกลุ่มฟอสเฟต) ที่ได้รับการรายงานว่าสามารถยับยั้งเส้นทางการส่งสัญญาณ BMP และ TGF-βได้
การศึกษานี้ตีพิมพ์ใน วารสาร Biochemical and Biophysical Research Communications
ศาสตราจารย์ฮายาตะกล่าวว่า: "RANKL ทำหน้าที่เป็น 'คันเร่ง' สำหรับการสร้างความแตกต่างของเซลล์สร้างกระดูก การขับรถไม่เพียงต้องใช้คันเร่งเท่านั้น แต่ยังต้องใช้เบรกด้วย ที่นี่เราพบว่า Ctdnep1 ทำหน้าที่เป็น 'เบรก' ในการสร้างความแตกต่างของเซลล์สร้างกระดูก" หน้า>
นักวิจัยได้ตรวจสอบการแสดงออกของ Ctdnep1 ในมาโครฟาจจากหนูที่ได้รับการรักษาด้วย RANKL และเซลล์ควบคุมโดยไม่ต้องได้รับการรักษาเป็นครั้งแรก พวกเขาตั้งข้อสังเกตว่าการแสดงออกของ Ctdnep1 ไม่เปลี่ยนแปลงเพื่อตอบสนองต่อการกระตุ้น RANKL อย่างไรก็ตาม มีการแปลเป็นภาษาท้องถิ่นในไซโตพลาสซึมในรูปแบบเม็ดเล็กในมาโครฟาจและสร้างความแตกต่างเป็นเซลล์สร้างกระดูก แตกต่างจากการแปลเป็นภาษาท้องถิ่นในนิวเคลียสปกติในเซลล์ประเภทอื่น โดยบ่งบอกถึงการทำงานของไซโตพลาสซึมในการสร้างความแตกต่างของเซลล์สร้างกระดูก
นอกจากนี้ Ctdnep1 การทำให้ล้มลง (การควบคุมยีนที่ลดลง) ส่งผลให้จำนวนเซลล์สร้างกระดูกที่เป็นบวกเพิ่มขึ้นสำหรับกรดฟอสฟาเตสที่ทนต่อทาร์เทรต (TRAP) โดยที่ TRAP เป็นเครื่องหมายของเซลล์สร้างกระดูกที่แตกต่าง
การทำให้ล้มลงด้วย Ctdnep1 ส่งผลให้มีการแสดงออกที่เพิ่มขึ้นของเครื่องหมายสร้างความแตกต่างที่สำคัญ ซึ่งรวมถึง "Nfatc1" ซึ่งเป็นปัจจัยการถอดรหัสหลักที่เกิดจาก RANKL สำหรับการสร้างความแตกต่างของเซลล์สร้างกระดูก ผลลัพธ์เหล่านี้สนับสนุน "ฟังก์ชันยับยั้ง" ของ Ctdnep1 โดยจะควบคุมการสร้างความแตกต่างของกระดูกในเชิงลบ นอกจากนี้ การล้มลงของ Ctdnep1 ยังส่งผลให้การดูดซึมแคลเซียมฟอสเฟตเพิ่มขึ้น ซึ่งบ่งชี้ถึงบทบาทในการยับยั้ง Ctdnep1 ในการสลายของกระดูก
ในที่สุด แม้ว่าการทำให้ล้มลงของ Ctdnep1 ไม่ได้เปลี่ยนเส้นทางการส่งสัญญาณ BMP และ TGF-β แต่เซลล์ที่ขาด Ctdnep1 ก็แสดงระดับโปรตีนฟอสโฟรีเลต (กระตุ้น) เพิ่มขึ้นซึ่งเป็นผลิตภัณฑ์ของเส้นทางการส่งสัญญาณ RANKL ผลลัพธ์เหล่านี้ชี้ให้เห็นว่าผลการปราบปรามของ Ctdnep1 ต่อการสร้างความแตกต่างของเซลล์สร้างกระดูกอาจไม่สามารถเป็นสื่อกลางผ่านเส้นทางการส่งสัญญาณ BMP และ TGF-β แต่ผ่านการควบคุมเชิงลบของเส้นทางการส่งสัญญาณ RANKL และระดับโปรตีน Nfatc1
โดยรวมแล้ว ผลลัพธ์เหล่านี้ให้ข้อมูลเชิงลึกใหม่เกี่ยวกับกระบวนการสร้างความแตกต่างของเซลล์สร้างกระดูก และระบุเป้าหมายการรักษาที่เป็นไปได้ ซึ่งสามารถนำมาใช้ในการพัฒนาการรักษาที่มีเป้าหมายในการลดการสูญเสียมวลกระดูกเนื่องจากการทำงานของเซลล์สร้างกระดูกมากเกินไป นอกจากโรคที่เกิดจากการสูญเสียกระดูกแล้ว Ctdnep1 ยังถูกระบุว่าเป็นปัจจัยเชิงสาเหตุใน medulloblastoma ซึ่งเป็นเนื้องอกในสมองในวัยเด็ก ผู้เขียนมองในแง่ดีว่าการวิจัยสามารถขยายไปสู่โรคอื่นๆ ในมนุษย์ได้ นอกเหนือจากการเผาผลาญของกระดูก
ศาสตราจารย์ฮายาตะสรุป: "ผลลัพธ์ของเราชี้ให้เห็นว่าจำเป็นต้องใช้ Ctdnep1 เพื่อป้องกันการสร้างเซลล์สร้างกระดูกมากเกินไป ผลลัพธ์เหล่านี้อาจขยายความรู้เพิ่มเติมว่าเครือข่ายฟอสโฟรีเลชั่น-ดีฟอสโฟรีเลชั่นควบคุมการสร้างความแตกต่างของเซลล์สร้างกระดูกอย่างไร และอาจให้กลยุทธ์การรักษาใหม่ๆ สำหรับการรักษาโรคกระดูกที่เกี่ยวข้องกับ กิจกรรมของกระดูกที่มากเกินไป"